Von Hippel Lindau Disease
By Paul T. Finger, MD
Description
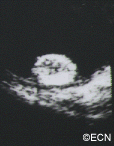
Von Hippel-Lindau (VHL) disease is an autosomal dominantly inherited multisystem cancer syndrome with a predilection for the central nervous system (CNS) and the retina. Retinal capillary “Vvon Hippel” hemangioma is one of the most common and often the earliest manifestations of VHL disease and, therefore, ophthalmologists are frequently involved in the care of patients with this disease. The incidence of VHL disease is approximately one in 40,000 live births and it is estimated that there are approximately 7,000 patients with VHL disease in the United States.
Symptoms
Ophthalmic Findings:
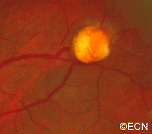
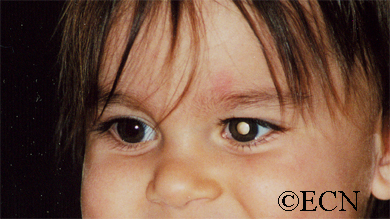
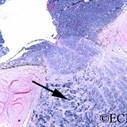
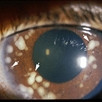
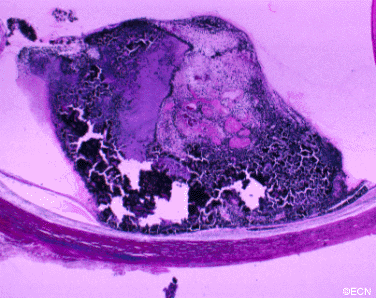
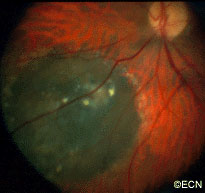
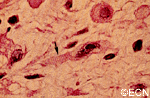
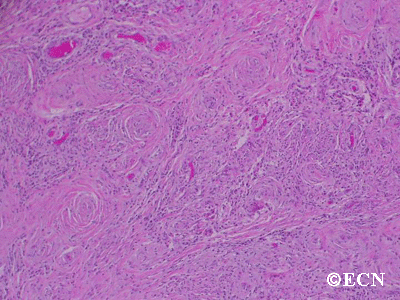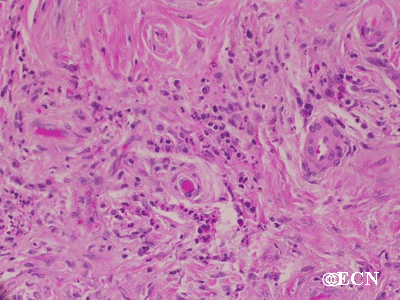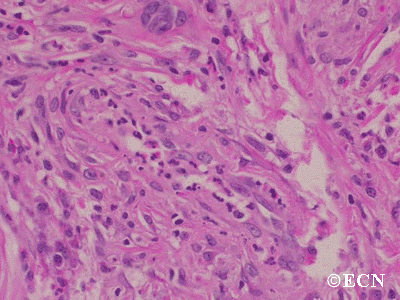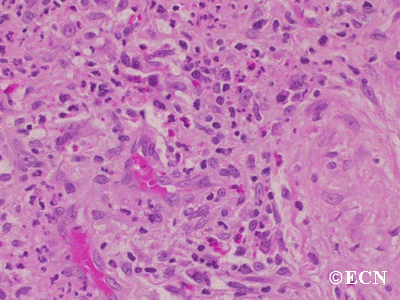
The main ophthalmic finding in VHL is retinal capillary hemangioma, which is a benign hamartoma. The anterior segment can be secondarily involved due to complications such as neovascular glaucoma and cataract formation. A large cohort study found only 2% of eyes had neovascularization of the iris. If a patient has a solitary retinal capillary hemangioma, they do not necessarily have VHL disease. However, they should undergo genetic testing.
Diagnosis
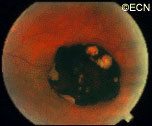
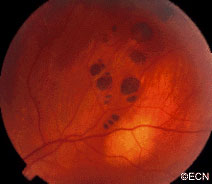
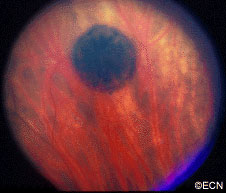
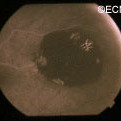
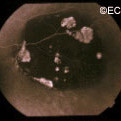
Retinal capillary hemangiomas are usually orange red, circumscribed, round, vascular tumors supplied by a pair of dilated and tortuous feeder vessels. They are most commonly located in the temporal peripheral retina. Juxtapapillary retinal capillary hemangiomas are less common, representing about 11% to 15% of cases, and their appearance can vary depending on whether the lesion is endophytic (grow from the retina into the vitreous gel of the eye), exophytic (grow beneath the retina), or sessile (flat).
Retinal capillary hemangioma usually manifests as a solitary tumor, but approximately one-third of patients have multiple retinal hemangiomas, and up to half of patients have bilateral involvement.
Secondary effects (from the retinal angioma) leading to visual loss, such as intraretinal and subretinal exudation, are often limited to the vicinity of the hemangioma but can be remote, often producing a macular star of exudates. Glial proliferation can lead to tractional retinal detachment and macular pucker. Retinal or vitreal hemorrhages are rarely observed, occurring in fewer than 3% of cases.
Frequency and Patient Related Findings:
The frequency of occurrence of retinal capillary hemangiomas in VHL disease has been
reported to vary from 49% to 85%. The mean age at diagnosis of retinal capillary hemangioma in VHL disease is approximately 25 years, and most patients present between the ages of 10 and 40 years. The probability of developing a retinal capillary hemangiomas increases progressively with age. Recent publications indicate that the hemangioma is usually manifested by age 30, and the prevalence rate is stable thereafter. Therefore adults with a normal retina at age 30 years may have a low risk of developing a retinal capillary hemangioma during the reminder of their lives. The natural course of retinal capillary hemangiomas is variable (progression, stability or spontaneous regression). Small lesions may remain stable for years or may show evidence of gliosis without leakage, but some have been documented to enlarge. Most hemangiomas, however, tend to enlarge progressively and lead to retinal changes. In late stages they may cause massive exudation and retinal detachment, uveitis (inflammation), glaucoma and phthisis (shrinking of the eye). Classification systems to aid in staging the clinical progression have been developed.
Other Tumors That May Look Like Von Hippel Angiomas:
The fundus findings of retinal capillary hemangioma are usually typical, and diagnosis can be made based on ophthalmoscopic examination. The diagnosis might be confused with retinal macroaneurysm or adult Coats disease when severe exudation exists.
Treatments
For treatment of Von Hippel retinal angiomas, see Conditions (Von Hippel Angioma) However, in ths section we will discuss the diagnosis of systemic disease and genetics:
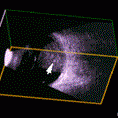
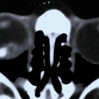
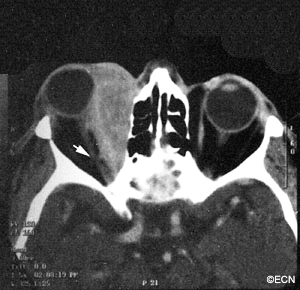
The systemic manifestations of VHL are multiple and include CNS hemangiomas of the brain and spinal cord, renal cell carcinomas, renal cysts, pheochromocytomas, pancreatic cysts, islet cell tumors, epididymal cystadenomas, endolymphatic sac tumors of the inner ear, and adnexal papillary cystadenomas of the broad ligament. After retinal capillary hemangioma, the most frequently affected organ systems are the CNS, kidneys and adrenal glands, many of them occurring years after the initial presentation with retinal capillary hemangiomas.
The diagnosis of VHL disease is based on three elements which include:
- retinal capillary hemangioma or CNS hemangioma
- visceral lesions
- family history of similar lesions
Surveillance:
After diagnosis is made, screening protocols should be followed, including urinary catecholamines and ophthalmoscopy on an annual basis with MRI of the brain and spinal cord every 2 to 3 years, and yearly abdominal US with an additional abdominal CT scan every 2 to 3 years.
Genetic Testing:
VHL disease is an autosomal dominant disease whose gene is located on chromosome 3p 25-26. The gene functions as a tumor suppressor gene that promotes tumor formation when its function is lost. The normal protein product of the VHL gene forms a complex with other proteins that targets hypoxia inducible factors (HIFs) for degradation. Mutations in the VHL gene result in stabilization of the HIFs, which bind to specific enhancer elements in the VEGF gene and stimulate angiogenesis. With a near-complete penetrance of the disease and only rare instances of mosaicism, genetic testing has been proved helpful in early diagnosis and clinical screening for disease manifestations.
SUMMARY
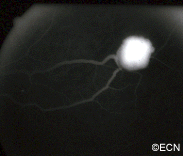
VHL disease is an autosomal dominantly inherited multisystem cancer syndrome with a predilection for the central nervous system and the retina. Retinal capillary hemangioma is one of the most common and earliest manifestations of VHL disease. Fundus findings are usually typical, and diagnosis can be made based on ophthalmoscopic examination, but fluorescein angiography is an additional informative diagnostic tool. Various treatment modalities exist, although the mainstays of therapy are laser photocoagulation and cryotherapy. VHL disease, however, is associated with significant mortality secondary to either CNS hemangioma or renal cell carcinoma. Life expectancy of affected individuals can be improved by early detection, genetic testing and systemic treatment. Treatment of this syndrome requires cooperation of physicians from multiple specialists, including those to treat the central nervous system and kidney tumors.
References
- Annesly WJ, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:446-453.
- Choyke PL, Glenn GM, Walther et al. The natural history of renal lesions in von Hippel- Lindau disease: a serial CT study in 28 patients. Am J Roentgenol. 1992;159:1229-1234.
- Dahr SS, Cusick M, Roudriguez-Coleman H, et al. Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina. 2007;27:150-158.
- Harris AL. von Hippel-Lindau syndrome. Target for anti-vascular endothelial growth factor (VEGF) receptor therapy. The Oncologist. 2000;5(suppl):32-36.
- Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943-944.
- Magee MA, Kroll AJ, Lou PL, Ryan EA. Retinal capillary hemangiomas and von Hippel-Lindau disease. Semin Opthalmol. 2006;21:143-150.
- Maher ER, Yates JR, Harries, et al. Clinical features and natural history of von Hippel-Lindau disease. QJM. 1990;77:1151-1163.
- Maher ER, Lselius L, Yates JR, Et al. von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443-447.
- Shields CL, Shields JA, Barret J, et al. Vasoproliferative tumors of the ocular fundus. Classification and clinical manifestations in 103 patients. Arch Ophthalmol. 1995;113:615-623.
- Sigelman J. Retinal diseases. Pathogenesis, laser therapy and surgery. Boston:Little Brown and Company. 1984:316.
- Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117-142.
- Vail D. Angiomatosis retinae, eleven years after diathermy coagulation. Am J Ophthalmol. 1958;46:525-534.
- Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371-378.
- Wong WT, Chew EY. Ocular von Hippel-Lindau disease: clinical update and emerging treatments. Curr Opin Ophthalmol. 2008;19:213-217.
- Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology. 2008;115:1957-1964.