Retinoblastoma is the most common intraocular cancer of childhood and affects approximately 300 children in the United States each year. More than 96% of children in North America and Europe are cured of retinoblastoma due to early detection and treatment of the affected eye. This is not true for children in countries that do not have eye cancer specialists.
Unfortunately, some children can have both eyes affected. Whenever possible, eye-cancer specialists try to save a child’s eye and preserve their vision.
Symptoms
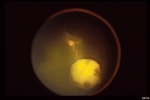
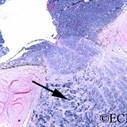
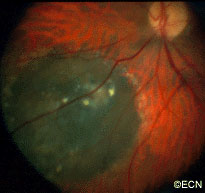
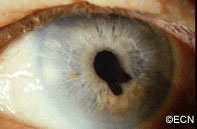
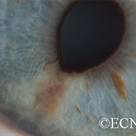
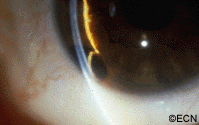
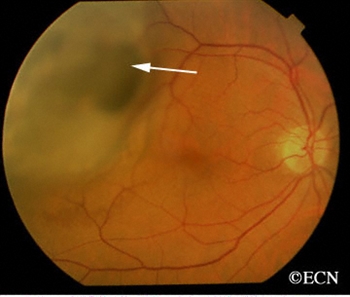
An intraocular photograph of an isolated “endophytic” retinoblastoma.
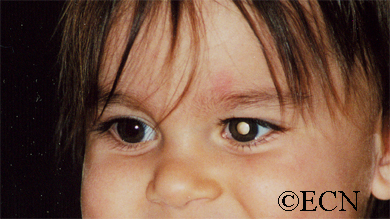
Leukocoria (white pupil) and misaligned eyes (strabismus) are the most common signs of retinoblastoma. In other cases, the child may have developed neovascular glaucoma and may be in pain. Longstanding glaucoma can cause enlargement of the eye (buphthalmos). Children with neovascular glaucoma and enlargement of the eye are at greater risk for extraocular spread of their retinoblastoma.
A family history of retinoblastoma can be very important. Retinoblastoma was the first cancer to be directly associated with a genetic abnormality (Deletions or mutation of the q14 band of chromosome 13). Retinoblastoma can occur sporadically (without a family history) or it can be inherited (with a family history).
If a genetic mutation is found there is a 45-50% chance that the parents will have another child with retinoblastoma. If there is no family history and no mutation is found, the risk of having a second child with retinoblastoma is 2-5%. The average age of children first diagnosed with retinoblastoma is 18 months (typical range 0 to 36 months).
Diagnosis
More than 75% of children with retinoblastoma are first noted to have a “white-pupil” (which the doctors call leukocoria), or poorly aligned eyes (which the doctors call strabismus), or a red and painful eye (usually due to glaucoma). Other eye diseases which can cause these symptoms include congenital cataract, Toxocara canis, Coat’s disease, and persistent hypertrophic primary vitreous (PHPV). These diseases may look like retinoblastoma, but by performing an eye examination under anesthesia (EUA), specialized blood tests, digital photography, radiographic scans, and ultrasound evaluations ophthalmic oncologists can diagnose intraocular retinoblastoma in over 95% of cases. In order to be 100% correct all the time, eye-cancer specialists would have to perform a biopsy. Biopsies of intraocular retinoblastoma are avoided in order to prevent cancer cells from spreading outside of the eye.
The presence of orbital extension, uveal involvement, and optic nerve invasion are known risk factors for the development of metastatic retinoblastoma.
Treatments
Retinoblastoma treatment typically requires the cooperation of an ophthalmic oncologist, pediatric oncologist, and radiation therapist. Over the last 30 years, treatment has evolved from simple enucleation (removal of the eye), to eye-sparing radiotherapy, and more recently to chemotherapy-based multi-modality therapy (for selected cases). Intra-arterial chemotherapy (IAC) has recently been investigated to save eyes, vision and spare the child from systemic chemotherapy.
Though retinoblastoma has been cured by external beam irradiation, investigators have found that radiation may cause an increase in the risk of developing second cancers later in life.
Protocols are currently being evaluated to use chemotherapy to shrink the retinoblastoma in order to treat them with laser therapy, freezing therapy (cryotherapy), and local “plaque” radiation. Where applicable, these techniques are thought to be safer than external beam irradiation for retinoblastoma. Intra-arterial chemotherapy is a newer method of perfusing the eye with chemotherapy, used for selected cases.
Treatment of retinoblastoma often requires a team of doctors made up of ophthalmic, radiation and pediatric oncologists. These doctors should evaluate your child, discuss all the different forms of treatment, and make them available.
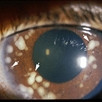
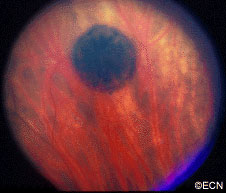
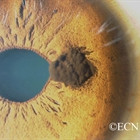
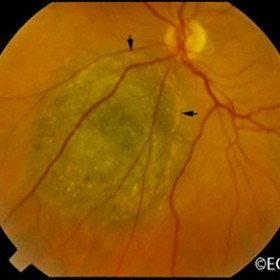
Note the flat, black well circumscribed lesion with areas of retinal pigment epithelial atrophy.
The retinal pigment epithelium (RPE) is a pigmented layer of the retina which can be thicker than normal at birth (congenital) or may thicken later in life. Areas of retinal pigment epithelial (RPE) hypertrophy usually do not cause symptoms. They are typically found during routine eye examinations.
Congenital retinal pigment epithelial hypertrophy (CHRPE) is usually found before patients reach 30 years of age. They may enlarge with time, but are not malignant. CHPRE has been an association with Gardner’s Syndrome (familial colonic polyposis).
This is a case of congenital hypertrophy of the retinal pigment epithelium, “bear-tracks.”
Therefore, if your eye care specialist has told you that you have CHPRE( pronounced CHER PEE), it is reasonable to tell your primary care physician or gastroenterologist (if you have one) so that he or she may recommend the frequency of colon screening tests.
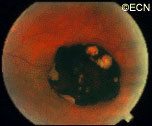
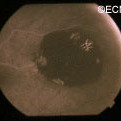
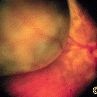
In contrast, acquired retinal pigment epithelial hypertrophy (RPEH) is typically found later in life. They are typically jet-black to gray, flat, with a halo around its edges. Variable in size, RPEH lesions may develop lacunae of lightly colored areas of atrophy (see image above). These
This area of retinal pigment epithelial hypertrophy demonstrates a blue hue.
tumors are more commonly found in the peripheral retina where thickness is more difficult to judge by ophthalmoscopy.
Symptoms
Almost all patients with retinal pigment epithelial hypertrophy do not have symptoms. These pigmented intraocular lesions are found by eye care specialists during dilated examination of the inside of the eye (ophthalmoscopy). Eye tumor specialists can typically differentiate between retinal pigment epithelial hypertrophy and melanoma by clinical examination (without a biopsy).
Diagnosis
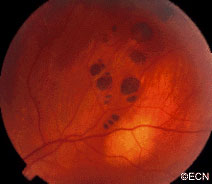
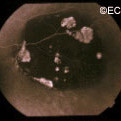
A small area of retinal pigment epithelial hypertrophy. It appears well circumscribed and with areas of relative lucency at the edges.
Retinal pigment epithelial hypertrophy (CHRPE and RPEH) can be diagnosed by ophthalmic examination. The eye examination will concentrate on the appearance of the retinal pigment epithelial hypertrophy. RPEH lesions tend to be black or atrophic. They may be surrounded by a halo of less pigmented tissue or exhibit a sharp demarcation line.
Ultrasonography typically shows that RPE hypertrophy is flat to minimally elevated and slightly hyper-reflective.
Fluorescein angiography of RPE hypertrophy typically demonstrates blockage of fluorescence (except in the areas of atrophy which are hyperfluorescent).
Optical coherence tomography (OCT) of RPE hypertrophy will demonstrate both thickening and thinning. The overlying retina is thinned, the retinal pigment epithelium is both thickened or can be thinned. The underlying choroid is typically thinned.
Treatment
Photographic documentation of these lesions is recommended for future comparison. Ultrasonography and fluorescein angiography is typically used to differentiate RPE hypertrophy from uveal melanoma and certain rare intraocular tumors. Serial observation is warranted in that RPE hypertrophy can enlarge over time.
The iris is the colored part of the eye. It is made up of two layers. The outer “stroma” can be blue, hazel, green or brown. The back layer (the iris pigment epithelium) is always brown. Tumors can grow within, through and thus behind the iris.
Symptoms
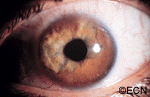
This tapioca-colored iris tumor has pulled the iris pigment epithelium out onto the surface of the iris stroma.
Iris melanoma patients usually have no symptoms. The tumor might be noticed by the patient, their family, or by the eye care specialist (during a routine eye examination). Some people have lots of freckles on their irides. Some of these pigmented spots have thickness and are called Nevi.
If the patient notices that one of their nevi has changed, enlarged or is pulling (ovalization) on the pupil; they should see an eye care specialist for evaluation and referral to an eye cancer specialist.
Diagnosis
Photographs of the surface of the iris tumor should be obtained to establish a baseline for
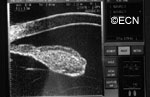
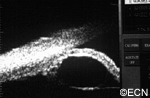
Ultrasound shows diffuse iris thickening of the iris stroma with bowing and invasion of the underlying iris pigment epithelium.
future comparisons. High frequency ultrasound is used to examine and measure the iris tumor. Ultrasound can reveal if the tumor is cystic or solid, how it extends within the iris and ciliary body. Ophthalmic oncologists use high frequency ultrasound measurements to evaluate iris tumors for evidence of growth or regression after treatment.
Characteristics that suggest that an iris tumor is cancerous include seeing blood vessels within the tumor (intrinsic vascularity), secondary glaucoma, evidence that the pupil is deformed (ectropion uveae), and the development of a cataract beneath the tumor. Some eyes may have enlarged “sentinel”blood vessels on the white of the eye (sclera) in the quadrant of the tumor.
The most important finding is documented growth. Since iris melanomas are commonly small, and less commonly (10-11%) spread to other parts of the body, these tumors are often watched for evidence of growth or change before biopsy or treatment is considered. A small amount of growth is not thought to significantly affect the rate of metastasis from a small iris melanoma. When necessary, eye cancer specialists can biopsy an iris tumor to help determine if the tumor is benign or malignant.
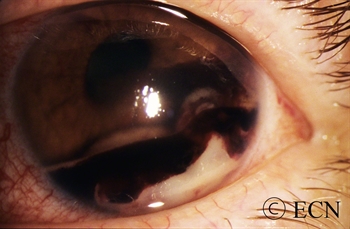
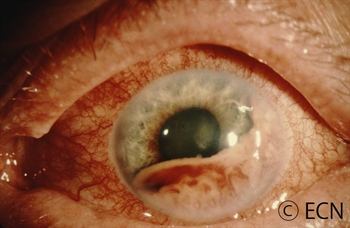
A diffuse iris melanoma causing severe glaucoma was treated by enucleation. At The New York Eye Cancer Center, we usually treat these cancers with eye and vision-sparing plaque radiation.
Treatments
Most pigmented iris tumors do not grow. They are photographed and monitored with periodic observation. When an iris melanoma is documented to grow, we know that it can damage the eye, cause secondary glaucoma and spread to other parts of the body. Then treatment risks become more acceptable to the patient. At The New York Eye Cancer Center, we typically use plaque radiation to destroy iris and iridociliary melanomas. This treatment allows for preservation of the iris, the pupil and does not cloud the cornea. We have found that the most common complication is cataract and permanent vision loss is rare.
Small Iris Melanomas:
Though most small iris melanomas can be surgically removed, however iridectomy may cause glare and astigmatism. The function of the iris and size of the pupil are better preserved if the tumor is destroyed with plaque radiation.
Medium-sized Iris Melanomas:
Though many of these tumors can be surgically removed, plaque or proton radiotherapy should be considered as primary treatment for these tumors. Though a radiation cataract is likely to develop, due to the distance between the radiation and the macular retina, vision limiting radiation retinopathy is very unlikely.
Large-sized Iris Melanomas with Advanced Glaucoma:
These cases can often be difficult to treat with either surgical removal or eye-sparing radiation therapy. Cure for these tumors is likely to require removal of the eye.
Diffuse Iris Melanomas:
Sometimes the entire iris is filled with melanoma. In these cases, removal of the eye is a reasonable option. However, there has been a recent trend towards and our experience that eye and vision-sparing radiation of the entire front of the eye (anterior segment) can be used to control the tumor, spare vision and allow the patient to keep their eye.
Melanocytoma is a form of nevus that can occur in and behind the iris in the ciliary body. In the image seen below, this melanocytoma has a cobblestone textured surface and feathered margins. Interestingly, it is also causing a small amount of pupillary distortion (correctopia). Bits of iris melanocytoma can break off and settle in the inferior angle. These sedimentary melanocytoma cells can clog the natural drain of the eye (trabecular meshwork), causing increased eye pressure (glaucoma). Melanocytoma tumors can grow, malignant transformation is rare and metastasis is reportable.
Symptoms
Most patients with iris melanocytoma can see a dark spot on their iris, and have no other symptoms. Patients can have pigment dispersion, secondary glaucoma and intraocular inflammation (iritis). Secondary glaucoma can either be asymptomatic, cause one-sided (ipsilateral) headaches and cloudy vision (with halos around lights).
Diagnosis
Iris melanocytoma can be diagnosed by clinical examination. The tumor tends to be dark brown to black and the edges feather-shaped. The surface can be cobblestone (bumpy) appearance or smooth.
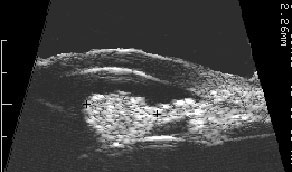
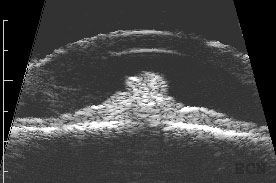
High-frequency ultrasound is particularly helpful for the diagnosis of iris melanocytoma. This technique is used to evaluate the depth of penetration into the iris and surrounding tissues. It can also be used to monitor for growth. These tumors can extend through the iris and into the ciliary body. Unlike low reflective iris melanomas, they tend to appear bright (highly reflective).
Longitudinally oriented 20 MHz high-frequency ultrasound demonstrates the club-shaped melanocytomaTransverse oriented 20 MHz high-frequency ultrasound demonstrates its cobblestone-shaped surface.
Treatments
Iris melanocytoma does not usually require treatment. High-quality slit-lamp and gonioscopic photographs should be taken to record the appearance and surface characteristics of the tumor. High-frequency ultrasound should be performed to evaluate its thickness and extension into surrounding structures. These evaluations will be used to monitor for growth. Careful attention should be made to measuring intraocular pressure. Secondary glaucoma can occur due to pigment that clogs the natural drain of the eye (trabecular meshwork). Patients should keep a copy of these baseline tests in case they move to another area or their doctors change.
Should an iris melanocytoma be found to grow, an iris biopsy can be performed to determine if it is benign or malignant. When possible, rapidly growing and malignant iris melanocytomas whould be surgically removed. If resection is not possible, they can be treated with relatively high-dose plaque radiation therapy.
Additional info
Secondary melanocytomalytic glaucoma can be treated like other glaucomas. It is reasonable to consult with a glaucoma specialist. Strenuous exercise may “shake-up” the intraocular pigment and cause a temporarily increased eye pressure. When this happens, patients may notice headaches or cloudy vision after exercise.
If glaucoma surgery is recommended (e.g. trabeculectomy, stent, valve), a tumor biopsy can ensure that the tumor is not a melanoma.
Cysts can form in different parts of the iris and ciliary body. Most remain undetected, unless they push on the iris or get relatively large. The most common is the neuro-epithelial iris cyst, which is typically located beneath the iris root. Cysts can also be located in the ciliary body, the iris stroma, and be formed by splitting (schisis) of the iris pigment epithelium). These cysts can push the iris forward, appear as a mass or tumor and may cause angle closure glaucoma.
Symptoms
Almost all iris cysts are located behind the iris and cause no symptoms. They are found by the eye care specialist during ophthalmic examination. Iris stromal cysts can become visible on the surface of the iris. Most commonly, the eye doctor sees a bulge in the iris stroma and considers that a tumor may be pushing it forward. At that point, the doctor may send the patient for an ocular tumor evaluation (to look behind the iris with ultrasound or OCT imaging).
Diagnosis
Most iris cysts can be diagnosed by clinical examination with high-frequency ultrasonography.
Large cysts can be seen by routine slit-lamp examination.
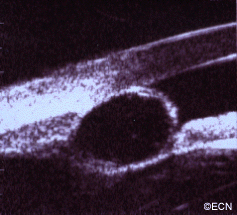
High frequency ultrasonography reveals internal echolucency consistent with the diagnosis of iris stromal cyst.This iris stromal cyst extends from the iris stroma onto the corneal endothelium.
Though cysts are benign, periodic observation is necessary due to the rare instances of secondary angle closure glaucoma (only seen with giant cysts, multifocal cysts and iris schisis cysts).
Slit-lamp photograph of an Iris Pigment Epithelial Cyst. Note its smooth, rounded surface.High-frequency ultra-sonography of a neuro-epithelial iris cyst. Note anterior displacement of the iris with focal angle closure.
High-frequency ultrasonography (aka UBM) has revolutionized our ability to image and thus “see” tissues behind the iris. It has allowed for evaluation of tumor thickness, depth of penetration, and invasion of adjacent tissues. Ultrasounds ability to determine if a tumor is solid or cystic has been particularly helpful.
Treatments
Most iris cysts do not require treatment.
Treatment is performed for the rare instances of secondary glaucoma or when the visual axis (pupil) becomes blocked. Laser has been used to deflate iris pigment epithelial cysts with minimal side effects.
The natural history of iris cysts has not been determined. In my experience, some get smaller, some get larger and most stay the same. Until eye care specialists understand which cysts will grow, periodic observation is warranted.
Intraocular metastasis is the most common malignant intraocular tumor and may occur in as many as 10% of patients with metastatic cancer. Most of these tumors go undetected unless they affect vision and become symptomatic.
Metastatic breast cancer is the most common in women and lung is the most common in men. Other primary sites include prostate, skin, kidney, colon and thyroid. Leukemia and lymphoma also occur in the eye.
Less than 10 percent of intraocular metastatic tumors are located in the anterior part of the eye.
Metastatic Lung Cancer
Symptoms
Most patients with iris metastasis present either with a visible cance (see photographs) or tumor-related “secondary” glaucoma. Glaucoma can cause foggy vision, halos around lights and headaches (brow aches). These “symptomatic” patients that come to or are sent for ophthalmic examination.
Most iris metastasis patients either have a history of cancer or are found to have a primary source of the tumor (on systemic examination). It is important to perform complete systemic survey when intraocular metastasis is suspected. This examination should include radiographic imaging (staging).
The New York Eye Cancer Center protocol involves whole body, scalp-to-toes PET/CT.
The reason is that though most intraocular cancers come from the lung and breast, there exist multiple less common “source” primary cancers. PET/CT evaluates the entire body. Consultation with an adult or pediatric medical oncologist should be obtained.
Despite systemic evalutions, sometimes patients will have no detectable primary cancer. Most of these tumors will later be found to originate from the lung (particularly in men). In these cases (where no primary cancer is found), the intraocular tumor becomes the only tissue that can be used to direct the search for its site of origin. Thus, biopsy of an anterior segment metastasis may become necessary. Biopsy techniques include: fine-needle aspiration biopsy, transcorneal tumor excision, and the relatively safe “Finger Iridectomy Technique (FIT).”
Treatments
Advanced anterior iris and ciliary body metastases can be difficult to manage. Radiation therapy alone or in combination with intraocular anti-VEGF drug therapy may be used to control the tumor, protect vision and conserve the eye. If left untreated, metastatic cancer in the iris can cause glaucoma and a blind painful eye. Thankfully, symptomatic anterior segment metastases are much less common than choroidal metastasis.
When the diagnosis of intraocular metastasis is made or suspected, eye cancer specialists always dilate and examine both eyes. This is because metastases can be both bilateral and multifocal. Computed tomography or magnetic resonance imaging of the brain and lungs should be performed due to a high incidence of concurrent metastases.
The wall of the eye has 3 main layers. From outside to inside there is: the white sclera, a blood vessel layer called the uvea (choroid, ciliary body and iris) and an inner retinal layer. Further, the pigment producing cells, “melanocytes” are primarily found in the vascular uveal layer. It is those melanocytes that can turn into malignant melanoma. Therefore, when melanoma happens in the choroid, they are called “choroidal melanoma,” the most common primary intraocular malignancy in adults. That said, choroidal melanomas are rare with 5 to10 out of each million people diagnosed with a choroidal melanoma each year. Choroidal melanomas can spread to other parts of the body.
Eye cancer specialists can determine if you have a choroidal melanoma by performing a complete eye examination with testing. This includes asking questions about your medical history, examining both of your eyes, looking into the eye through a dilated pupil, performing an ultrasound examination, and specialized photography (to examine the circulation within the choroidal melanoma).
MOST – Fingers’ Melanoma Mnemonic
Dr. Finger has developed the mnemonic device “MOST” to help eye care specialists to determine if the intraocular tumor is a melanoma.
“M,” Melanoma:
“O,” Orange Pigment Lipofuscin (OPL) a metabolic side product of cell death. This finding tells us that that either the underlying tumor is destroying the overlying tissue or itself is degenerating. Lipofuscin is best seen with a photographic test called Fundus Auto Fluorescence imaging, or FAF.
“S,” Subretinal fluid (SRF) is created by poorly formed or new, “neovascular” blood vessels within the tumor. Cancers need new vessels in order to grow. Large amounts of SRF can be seen by ophthalmoscopy (looking into the eye) and ultrasound imaging. However, small amounts of SRF are best seen on 3D optical coherence tomographic imaging (3D-OCT).
“T,” Thickness of the tumor has been associated with malignancy. Simply, the thicker it is the more likely a pigmented intraocular tumor is malignant. It is widely accepted that tumors greater than 2.0 mm are more likely to be malignant. Ultrasound imaging is currently the best method to measure tumor thickness.
Your specialist will also request that you have a complete general medical check up and specific tests depending upon what they see inside your eye. In the Collaborative Ocular Melanoma Study (COMS), participating eye cancer specialists correctly diagnosed select choroidal melanoma in over 99.6% of cases (without a biopsy). That said, patients with unusual appearing “atypical” tumors were not entered into the study.
Classic Indications for Biopsy
Atypical tumor, metastatic tumor with no observable primary cancer and when the patient requests a pathology diagnosis. More recently, primarily due to genetic testing services, more and more centers are routinely performing choroidal tumor biopsy primarily for genetic tumor analysis. Genetics offers information about the tumor, but does not allow doctors to avoid treatment or follow up systemic testing for metastasis.
Choroidal biopsy has been associated with a risk of hemorrhage, infection, retinal detachment and a poorly quantified risk of tumor seeding (outside the eye). Risks related to tumor seeding are thought to be small, but clearly they have not been evaluated by any large prospective or retrospective study. Each eye cancer specialist should discuss the relative risks (known and unknown) of biopsy prior to surgery.
Questions About Intraocular Biopsy:
Remove the need for surgical tumor treatment?
Reduce the number of radiologic examination or years needed for systemic surveillance?
What are the risks of biopsy (hemorrhage (e.g. vitreous, subretinal, subfoveal), seeding, damage to the lens, optic nerve, retinal detachment, cataract, epiretinal membrane, loss of vision, loss of eye and/or reaction to anesthesia).
Symptoms
Most choroidal melanoma patients have no symptoms. The melanoma is found on routine eye examination. If patients have choroidal melanoma symptoms, they are usually seeing “flashes of light,” noticing “distortion” or loss of vision, and floating objects (floaters) in their vision.
If the choroidal melanoma is in the front of the eye (near the natural lens), it can push or tilt the natural lens causing an irregular astigmatism (blurring of vision).
Choroidal melanoma can leak fluid beneath the retina, making the retina detach and cause symptoms of flashing lights and floating specks “floaters”.
If the choroidal melanoma is in the macula (center of vision), it can grow beneath the fovea making the patient far-sighted. The choroidal melanoma can also grow into and destroy the fovea causing distortion, loss of vision or changes in color perception.
It is important to note that most patients with choroidal melanoma have no symptoms at all. Their tumors are found when they visit their eye doctor for a “routine” eye examination. So everyone should have periodic eye examinations (including dilated ophthalmoscopy). In general, the earlier and smaller the choroidal melanoma, the better the prognosis for both life and sight.
Other, more unusual presentations of anterior choroidal and iridociliary melanoma are discoloration of the iris, a brown spot on the outside of the eye, an irregularly shaped pupil and glaucoma.
Diagnosis
Choroidal melanoma is usually seen by ophthalmoscopy (when your eye doctor looks through a lens into your dilated pupil). Choroidal melanoma has typical “diagnostic” characteristics that include but are not limited to: pigmentation, low to moderate internal ultrasound reflectivity, clumps of orange pigment lipofuscin on its surface, leakage of subretinal fluid, or retinal detachment (on or around the choroidal melanoma) and thickness.
Pigmentation is due to naturally occurring melanin that comes from melanocyte cells in the choroidal layer of the eye. Choroidal melanomas are most commonly pigmented, but can be variably pigmented and even non-pigmented (amelanotic). Non-pigmented choroidal melanoma is due to a proliferation of melanocytes that have lost their ability to make the melanin pigment.
Orange pigment is made up of a chemical called lipofuscin and appears on the surface of choroidal melanomas. Lipofuscin is a product of cell death which indicate that cells are dying on the tumor’s surface. This is also sign of metabolic activity. Melanomas are more metabolically active than choroidal nevi.
Ultrasound is typically used to measure the choroidal melanoma size, evaluate internal tumor reflectivity, and look for melanoma extension behind the eye into the orbit called extrascleral extension. Ultrasound imaging has demonstrated that most choroidal melanomas are shaped like a dome and less commonly like a mushroom. Ultrasound can also evaluate and detect choroidal melanoma associated retinal detachment. However, optical coherence tomography (OCT) is a more sensitive way to detect subretinal fluid – retinal detachment.
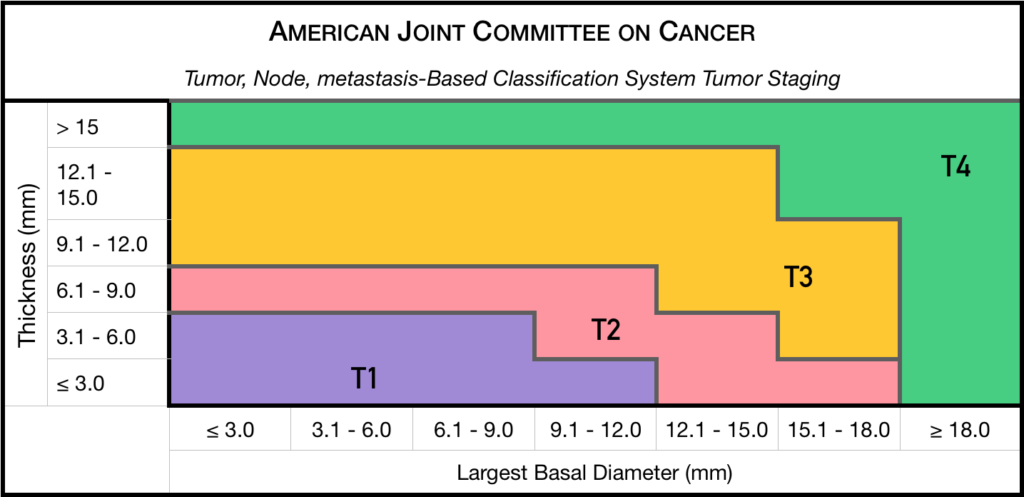
Staging
Chaired by Dr. Paul T. Finger, a committee of top ophthalmic specialists assembled to form the AJCC-UICC Ophthalmic Oncology Task Force. To ensure a broad range of specialists, Dr. Finger “internationalized” the committee, including over 58 members both from the USA and around the world.
This committee had one driving goal: to design a clinically useful Tumor-Node-Metastasis (TNM) based classification “language” for ocular tumors. This first-of-its-kind classification system has become a universal language for all who diagnose and treat ocular tumors.
Not only does a universal classification system offer cancer staging for the patient, it also allows physicians to directly compare data. In the long run, a common “eye tumor” language helps usdetermine and differentiate treatment types as well as coordinate the efforts of researchers working for a cure.
Dr. Finger has since translated this staging system for the worlds’ Union International for Cancer Control (UICC), and offered it to the world. In order to get everyone to employ this new language, he has recruited all the major medical journals to require eye cancer researchers to use AJCC-UICC staging.
Treatments
Small Choroidal Melanoma (AJCC T1 and T2): Patients with a small choroidal melanoma can be treated after their first visit, but since growth helps to prove that the tumor is a cancer, your doctor may suggest “observation” or watching for a small amount of choroidal melanoma growth prior to treatment. Your eye cancer specialist should discuss the relative risks and potential benefits of “observation for growth” as compared to “immediate treatment” for choroidal melanoma. If growth is documented (typically within 6 months of observation), eye cancer specialists will typically recommend definitive treatment.
Medium-sized Choroidal Melanoma (AJCC T3 and T4): Most patients with large-sized choroidal melanoma can be also be treated with eye-sparing low energy radiation therapy (e.g. palladium-103). However, larger tumors require more radiation and larger irradiated intraocular volumes resulting in greater risk of radiation side-effects and poor vision. Rarely such eyes have to be secondarily removed. Eye cancer specialists try to preserve eyes, even if the eye had reduced vision.
Large-sized Choroidal Melanoma: Very large choroidal melanomas (greater than 22 mm width) may be treated by initial removal of the eye (enucleation). This is because the amount of radiation required to destroy a choroidal melanoma that fills most of the eye will likely be too much for the eye to tolerate.
However, most patients, even with very large-sized choroidal melanoma can be treated with eye-sparing radiation therapy. However, after eye sparing radiation for very large choroidal melanomas, eyes are at greater risk to have poor vision, secondary inflammation and may require secondary removal at a later date.
Additional Info
Patients often ask why they have a choroidal melanoma. While there is no one cause, choroidal melanoma is more common among patients with blue vs. brown eyes, those with outdoor occupations, and in Australia where there is a hole in the ozone layer. Therefore, though this hypothesis has yet to be proven, it seems reasonable to assume that choroidal melanoma is related to sunlight (ultraviolet exposure).
In that sunlight exposure has been linked to several eye cancers and diseases of the eye, Dr. Finger suggests that you “Think of Sunglasses as Sun Block for your Eyes” ™ and start wearing your UV blocking sunglasses. They make great gifts too!
Dr. Finger also often gets questions related to stage and spread. These two things are closely linked––choroidal melanoma size is most closely related to its risk for spread to other parts of the body (metastasis). In three separate studies, cumulatively involving almost 20,000 patients, the average rate of metastasis has been 50%. However, patients with smaller tumors have much lower rates compared to larger tumors. Therefore, patients should ask their eye cancer specialist about their tumors AJCC-UICC tumor size and risk for metastasis. In general, the larger the choroidal melanoma the worse the prognosis for both vision and metastasis.
"Very well treated by Dr. Finger. He explained everything I needed to know about my issue with detail and attention, putting me at ease and giving me confidence to handle this problem for the rest of my life.”
– N.N.