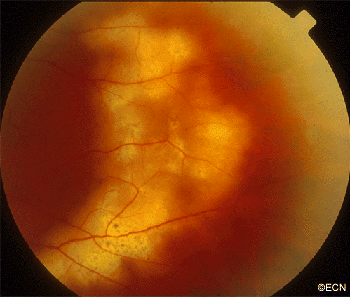
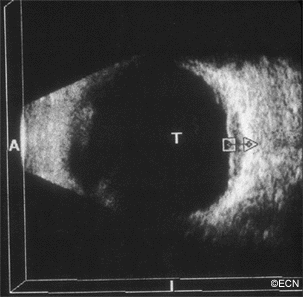
Malignant tumors from other parts of the body can spread in and around the eye. These tumors may never be discovered unless they affect the vision, are visible in the iris, or push the eye forward.
Symptoms
Most patients with choroidal metastasis have no symptoms, that is unless the tumor affects the central macular retina, optic nerve or the front of the eye (iris). For example, this patient’s tumor (pictured above) affected his optic nerve, caused decreased vision and floaters (spots in his vision).
Diagnosis
Most patients have a history of primary cancer (as in this case), and the tumor’s characteristics are typical of metastatic disease. This tumor was yellow-white, poorly circumscribed, less than 3 mm thick and causing congestion with vascular compromise of the optic nerve.
Treatments
As with this patient, most choroidal metastasis will resolve with external beam radiation therapy. Like many patients treated for metastatic disease, this patient received 30 Gy in 10 daily fractions. Other treatment protocols (doses) may be different, depending on the type of primary tumor.
Dr. Finger currently recommends that orbital radiation therapy for metastatic disease be performed in 180-200 cGy daily fractions. This is due to the unique sensitivity of the eye and lacrimal (tear) systems. With improvements in systemic therapy, patients are living longer and thus developing late radiation complications. Dr. Finger believes these complications will be less likely if the radiation is delivered in smaller daily fractions.
Choroidal melanoma can grow near, touch and even cover the optic nerve. Like other choroidal melanomas, they can exhibit orange pigment on its surface, subretinal fluid (localized retinal detachment), and thickness. The choroidal melanoma in the next photograph exhibits all three findings.
Symptoms
Juxtapapillary choroidal melanoma is typically near the central macular retina and may cause symptoms. Unlike most patients with choroidal melanoma, when the optic nerve is affected patients will have complaints of decreased vision, visual field defects, flashing lights or floaters (spots). However, most choroidal melanomas are found on routine eye examination with dilated ophthalmoscopy.
Diagnosis
Choroidal melanoma that affects the optic nerve must be differentiated from optic nerve melanocytoma. The eye care specialist will examine the tumor for evidence of orange pigment (lipofuscin, melanolipofuscin), thickness (as measured by ultrasound), for leakage (as measured by photography with angiography), and with optical coherence tomography (OCT).
Unlike optic nerve melanocytoma, malignant choroidal melanoma does not typically spread-out along the nerve fiber layer and are lighter in color. In addition, melanocytoma-OCT findings can be particularly helpful showing tumor cell invasion of the overlying retina and vitreous. In either case, it can be difficult to determine the diagnosis of very small tumors. In these cases, the tumor can be closely watched “followed” for evidence of growth, then a biopsy may need be performed. Dr. Finger has found that melanomas that completely encircle the optic disc (circumpapillary) are likely to cause an afferent pupillary defect.
Treatments
Choroidal melanoma that encircles or covers the optic nerve are particulary difficult to treat with eye-sparing plaque radiation therapy. This is because the optic nerve widens as it leaves the eye into the orbit. In addition, it becomes encased in the optic nerve sheath. Dr. Fingers’ 3D ultrasound studies showed that the nerve nearly triples in diameter behind the eye. Therefore, if a plaque is perfectly placed on the back of the eye, it can only reach to 1.5 mm from the optic disc (effectively missing the rest of the uncovered melanoma. This is why, many patients with melanomas touching or surrounding the optic disc were treated by removal (enucleation) of the affected eye.
Now with 12-years follow up, 94% of patients with choroidal melanoma do not have to lose their eye. For those tough to peripapillary, juxtapapillary and even circumpapillary melaoma, Dr. Finger invented 8-mm slotted plaques. The slot accomodated the entire optic nerve sheath into the plaque, thus allowing the plaque radiation to completely cover and surround the cancer. Fingers’ Slotted Plaques have been able to control more than 98% of these melanomas, has been able to spare some vision (with subsequent intensive anti-VEGF therapy) and allow patients to keep their eye. The patient must be counseled that they are at risk for impaired vision in the irradiated eye.
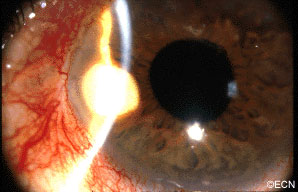
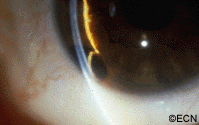
Slit-lamp photography shows the white pearl-cyst within the temporal iris stroma. In this case, a history of tumor growth and a mild iritis were noted.
A rare complication of cataract surgery, the “pearl-cyst” may grow from on and within the iris. The tumors are thought to be caused by displaced conjunctival or epidermal epithelium. Rupture of these mucous-containing cysts (within the eye) can cause severe glaucoma.
Symptoms
Most patients present due to ocular inflammation. They may notice the white tumor growing
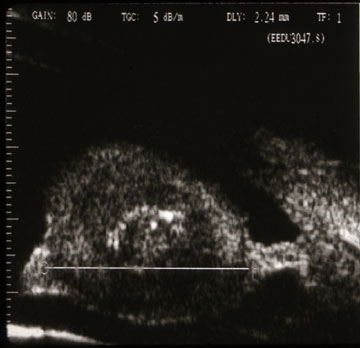
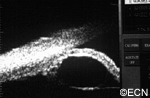
High frequency ultrasound demonstrates three layers (a mantle, mucus content, and cholesterol crystals at the center).
on the iris.
Diagnosis
The diagnosis of “Pearl Cyst” should be considered when a patient who has had intraocular surgery (cataract, corneal transplant, or filtering for glaucoma) presents with a slowly
enlarging white iris stromal tumor. High-frequency ultrasound imaging will show three layers: an outer mantle, a low reflective mucus
layer and a central, high-reflective cholesterol crystal core.
Treatments
When “Pearl-cyst” of the iris is suspected, complete surgical excision is warranted. Though difficult, the surgeon should try to remove the cyst intact. Should the cyst rupture during removal, the surgeon should be ready to aspirate or removal the cyst contents. The mucoid contents have been reported to cause secondary glaucoma. If the outer epithelial layer, the mantle, is left behind one runs the risk of cyst recurrence.
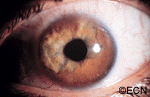
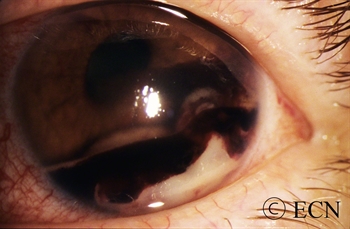
The iris is the colored part of the eye. It is made up of two layers. The outer “stroma” can be blue, hazel, green or brown. The back layer (the iris pigment epithelium) is always brown. Tumors can grow within, through and thus behind the iris.
Symptoms
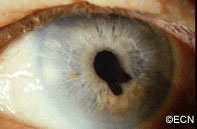
This tapioca-colored iris tumor has pulled the iris pigment epithelium out onto the surface of the iris stroma.
Iris melanoma patients usually have no symptoms. The tumor might be noticed by the patient, their family, or by the eye care specialist (during a routine eye examination). Some people have lots of freckles on their irides. Some of these pigmented spots have thickness and are called Nevi.
If the patient notices that one of their nevi has changed, enlarged or is pulling (ovalization) on the pupil; they should see an eye care specialist for evaluation and referral to an eye cancer specialist.
Diagnosis
Photographs of the surface of the iris tumor should be obtained to establish a baseline for
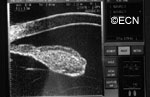
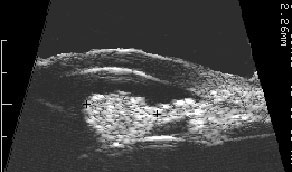
Ultrasound shows diffuse iris thickening of the iris stroma with bowing and invasion of the underlying iris pigment epithelium.
future comparisons. High frequency ultrasound is used to examine and measure the iris tumor. Ultrasound can reveal if the tumor is cystic or solid, how it extends within the iris and ciliary body. Ophthalmic oncologists use high frequency ultrasound measurements to evaluate iris tumors for evidence of growth or regression after treatment.
Characteristics that suggest that an iris tumor is cancerous include seeing blood vessels within the tumor (intrinsic vascularity), secondary glaucoma, evidence that the pupil is deformed (ectropion uveae), and the development of a cataract beneath the tumor. Some eyes may have enlarged “sentinel”blood vessels on the white of the eye (sclera) in the quadrant of the tumor.
The most important finding is documented growth. Since iris melanomas are commonly small, and less commonly (10-11%) spread to other parts of the body, these tumors are often watched for evidence of growth or change before biopsy or treatment is considered. A small amount of growth is not thought to significantly affect the rate of metastasis from a small iris melanoma. When necessary, eye cancer specialists can biopsy an iris tumor to help determine if the tumor is benign or malignant.
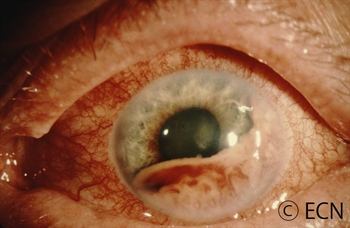
A diffuse iris melanoma causing severe glaucoma was treated by enucleation. At The New York Eye Cancer Center, we usually treat these cancers with eye and vision-sparing plaque radiation.
Treatments
Most pigmented iris tumors do not grow. They are photographed and monitored with periodic observation. When an iris melanoma is documented to grow, we know that it can damage the eye, cause secondary glaucoma and spread to other parts of the body. Then treatment risks become more acceptable to the patient. At The New York Eye Cancer Center, we typically use plaque radiation to destroy iris and iridociliary melanomas. This treatment allows for preservation of the iris, the pupil and does not cloud the cornea. We have found that the most common complication is cataract and permanent vision loss is rare.
Small Iris Melanomas:
Though most small iris melanomas can be surgically removed, however iridectomy may cause glare and astigmatism. The function of the iris and size of the pupil are better preserved if the tumor is destroyed with plaque radiation.
Medium-sized Iris Melanomas:
Though many of these tumors can be surgically removed, plaque or proton radiotherapy should be considered as primary treatment for these tumors. Though a radiation cataract is likely to develop, due to the distance between the radiation and the macular retina, vision limiting radiation retinopathy is very unlikely.
Large-sized Iris Melanomas with Advanced Glaucoma:
These cases can often be difficult to treat with either surgical removal or eye-sparing radiation therapy. Cure for these tumors is likely to require removal of the eye.
Diffuse Iris Melanomas:
Sometimes the entire iris is filled with melanoma. In these cases, removal of the eye is a reasonable option. However, there has been a recent trend towards and our experience that eye and vision-sparing radiation of the entire front of the eye (anterior segment) can be used to control the tumor, spare vision and allow the patient to keep their eye.
Melanocytoma is a form of nevus that can occur in and behind the iris in the ciliary body. In the image seen below, this melanocytoma has a cobblestone textured surface and feathered margins. Interestingly, it is also causing a small amount of pupillary distortion (correctopia). Bits of iris melanocytoma can break off and settle in the inferior angle. These sedimentary melanocytoma cells can clog the natural drain of the eye (trabecular meshwork), causing increased eye pressure (glaucoma). Melanocytoma tumors can grow, malignant transformation is rare and metastasis is reportable.
Symptoms
Most patients with iris melanocytoma can see a dark spot on their iris, and have no other symptoms. Patients can have pigment dispersion, secondary glaucoma and intraocular inflammation (iritis). Secondary glaucoma can either be asymptomatic, cause one-sided (ipsilateral) headaches and cloudy vision (with halos around lights).
Diagnosis
Iris melanocytoma can be diagnosed by clinical examination. The tumor tends to be dark brown to black and the edges feather-shaped. The surface can be cobblestone (bumpy) appearance or smooth.
High-frequency ultrasound is particularly helpful for the diagnosis of iris melanocytoma. This technique is used to evaluate the depth of penetration into the iris and surrounding tissues. It can also be used to monitor for growth. These tumors can extend through the iris and into the ciliary body. Unlike low reflective iris melanomas, they tend to appear bright (highly reflective).
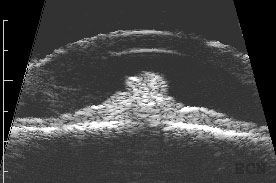
Longitudinally oriented 20 MHz high-frequency ultrasound demonstrates the club-shaped melanocytomaTransverse oriented 20 MHz high-frequency ultrasound demonstrates its cobblestone-shaped surface.
Treatments
Iris melanocytoma does not usually require treatment. High-quality slit-lamp and gonioscopic photographs should be taken to record the appearance and surface characteristics of the tumor. High-frequency ultrasound should be performed to evaluate its thickness and extension into surrounding structures. These evaluations will be used to monitor for growth. Careful attention should be made to measuring intraocular pressure. Secondary glaucoma can occur due to pigment that clogs the natural drain of the eye (trabecular meshwork). Patients should keep a copy of these baseline tests in case they move to another area or their doctors change.
Should an iris melanocytoma be found to grow, an iris biopsy can be performed to determine if it is benign or malignant. When possible, rapidly growing and malignant iris melanocytomas whould be surgically removed. If resection is not possible, they can be treated with relatively high-dose plaque radiation therapy.
Additional info
Secondary melanocytomalytic glaucoma can be treated like other glaucomas. It is reasonable to consult with a glaucoma specialist. Strenuous exercise may “shake-up” the intraocular pigment and cause a temporarily increased eye pressure. When this happens, patients may notice headaches or cloudy vision after exercise.
If glaucoma surgery is recommended (e.g. trabeculectomy, stent, valve), a tumor biopsy can ensure that the tumor is not a melanoma.
Cysts can form in different parts of the iris and ciliary body. Most remain undetected, unless they push on the iris or get relatively large. The most common is the neuro-epithelial iris cyst, which is typically located beneath the iris root. Cysts can also be located in the ciliary body, the iris stroma, and be formed by splitting (schisis) of the iris pigment epithelium). These cysts can push the iris forward, appear as a mass or tumor and may cause angle closure glaucoma.
Symptoms
Almost all iris cysts are located behind the iris and cause no symptoms. They are found by the eye care specialist during ophthalmic examination. Iris stromal cysts can become visible on the surface of the iris. Most commonly, the eye doctor sees a bulge in the iris stroma and considers that a tumor may be pushing it forward. At that point, the doctor may send the patient for an ocular tumor evaluation (to look behind the iris with ultrasound or OCT imaging).
Diagnosis
Most iris cysts can be diagnosed by clinical examination with high-frequency ultrasonography.
Large cysts can be seen by routine slit-lamp examination.
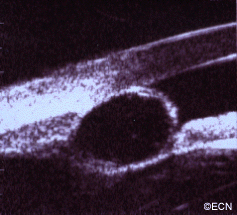
High frequency ultrasonography reveals internal echolucency consistent with the diagnosis of iris stromal cyst.This iris stromal cyst extends from the iris stroma onto the corneal endothelium.
Though cysts are benign, periodic observation is necessary due to the rare instances of secondary angle closure glaucoma (only seen with giant cysts, multifocal cysts and iris schisis cysts).
Slit-lamp photograph of an Iris Pigment Epithelial Cyst. Note its smooth, rounded surface.High-frequency ultra-sonography of a neuro-epithelial iris cyst. Note anterior displacement of the iris with focal angle closure.
High-frequency ultrasonography (aka UBM) has revolutionized our ability to image and thus “see” tissues behind the iris. It has allowed for evaluation of tumor thickness, depth of penetration, and invasion of adjacent tissues. Ultrasounds ability to determine if a tumor is solid or cystic has been particularly helpful.
Treatments
Most iris cysts do not require treatment.
Treatment is performed for the rare instances of secondary glaucoma or when the visual axis (pupil) becomes blocked. Laser has been used to deflate iris pigment epithelial cysts with minimal side effects.
The natural history of iris cysts has not been determined. In my experience, some get smaller, some get larger and most stay the same. Until eye care specialists understand which cysts will grow, periodic observation is warranted.
Intraocular metastasis is the most common malignant intraocular tumor and may occur in as many as 10% of patients with metastatic cancer. Most of these tumors go undetected unless they affect vision and become symptomatic.
Metastatic breast cancer is the most common in women and lung is the most common in men. Other primary sites include prostate, skin, kidney, colon and thyroid. Leukemia and lymphoma also occur in the eye.
Less than 10 percent of intraocular metastatic tumors are located in the anterior part of the eye.
Metastatic Lung Cancer
Symptoms
Most patients with iris metastasis present either with a visible cance (see photographs) or tumor-related “secondary” glaucoma. Glaucoma can cause foggy vision, halos around lights and headaches (brow aches). These “symptomatic” patients that come to or are sent for ophthalmic examination.
Most iris metastasis patients either have a history of cancer or are found to have a primary source of the tumor (on systemic examination). It is important to perform complete systemic survey when intraocular metastasis is suspected. This examination should include radiographic imaging (staging).
The New York Eye Cancer Center protocol involves whole body, scalp-to-toes PET/CT.
The reason is that though most intraocular cancers come from the lung and breast, there exist multiple less common “source” primary cancers. PET/CT evaluates the entire body. Consultation with an adult or pediatric medical oncologist should be obtained.
Despite systemic evalutions, sometimes patients will have no detectable primary cancer. Most of these tumors will later be found to originate from the lung (particularly in men). In these cases (where no primary cancer is found), the intraocular tumor becomes the only tissue that can be used to direct the search for its site of origin. Thus, biopsy of an anterior segment metastasis may become necessary. Biopsy techniques include: fine-needle aspiration biopsy, transcorneal tumor excision, and the relatively safe “Finger Iridectomy Technique (FIT).”
Treatments
Advanced anterior iris and ciliary body metastases can be difficult to manage. Radiation therapy alone or in combination with intraocular anti-VEGF drug therapy may be used to control the tumor, protect vision and conserve the eye. If left untreated, metastatic cancer in the iris can cause glaucoma and a blind painful eye. Thankfully, symptomatic anterior segment metastases are much less common than choroidal metastasis.
When the diagnosis of intraocular metastasis is made or suspected, eye cancer specialists always dilate and examine both eyes. This is because metastases can be both bilateral and multifocal. Computed tomography or magnetic resonance imaging of the brain and lungs should be performed due to a high incidence of concurrent metastases.
Most patients diagnosed to have intraocular lymphoma have symptoms of vitreous floaters, a history of systemic lymphoma or have been diagnosed as having chronic uveitis. Any patients with vitreous cells, no history of recent intraocular surgery and a non-painful eye should be suspected to have intraocular lymphoma.
Symptoms
Case Example: A 70-year-old woman was noted to experience an acute deterioration of her vision due to vitreous cells. A diagnostic vitrectomy was performed. Pathology showed large B-cell lymphoma. A complete metastatic survey (imaging studies of the brain, chest, abdomen), lumbar puncture and bone marrow biopsy were found to be negative.
Diagnosis
The diagnosis of intraocular lymphoma is typically made by removal of cells (vitrectomy biopsy) from the eye with subsequent cytopathologic evaluation. Lymphoma biopsy should be performed at a center with ophthalmic pathology services used to working with small specimens. Once the diagnosis is certain, a hematologist-oncologist should be consulted to perform an evaluation for systemic and central nervous system lymphoma (staging).
Treatments
B-scan ultrasound reveals an irregular retinal surface and variable internal reflectivity.
Though chemotherapy can be used to treat the systemic disease, poor intraocular drug penetration can leave residual lymphoma in the eye. In these cases, external beam radiation therapy to the eyes and orbits will typically clear the intraocular disease.
The literature consists of a multitude of case-reports and no evidence based comparative studies. In my experience, most patients are eventually treated with ocular irradiation. Others have had prophylactic whole-brain irradiation as well as combinations of local and systemic chemotherapy.
What is common to most of these reports is that, when the lymphoma is found in the brain, the prognosis is poor. Clearly, a natural history study followed by prospective randomized treatment trials would be helpful to find the cure for patients with intraocular lymphoma.
Additional info
Radiation Therapy
Radiation therapy has been the most common treatment for intraocular lymphoma. Both eyes are usually treated because approximately 80% of cases will either present as or go on to develop bilateral disease (within 8 years).
The prescription dose has been decreasing with less than 3,000 cGy being employed in many centers. The dose to the orbit is adjusted when whole-brain irradiation is required. With this in mind, remember to always obtain a complete neurological work-up to rule out central nervous system (CNS) involvement. Radiation will help acutely, but intraocular and CNS relapse are common.
Chemotherapy
Recent investigations have combined radiation therapy with systemic or intrathecal chemotherapy. Several studies suggest that this approach has prolonged survival. Chemotherapeutic agents have been given intravenously, intrathecally, and intravitreally (into the eye).
Injections of chemotherapy into the eye have been investigated as an alternative to radiation. Typically using methotrexate and/or retuximab, multiple injections are required and carry the known risks associated with ocular perforation (e.g. infection) as well as chemotherapy associated side-effects. This is why most centers use intravitreal chemotherapy when patients demonstrate intraocular recurrence after failure of radiation therapy.
In this case, our patient had a history of myeloma. If suspected, the work-up for multiple myeloma includes: a bone-marrow biopsy, skeletal x-rays, serum protein electrophoresis, urinalysis for Bence-Jones proteins. Several cases have described orbital involvement.
In this case, progression was documented and external beam radiation therapy stabilized her ocular condition.
"Very well treated by Dr. Finger. He explained everything I needed to know about my issue with detail and attention, putting me at ease and giving me confidence to handle this problem for the rest of my life.”
– N.N.


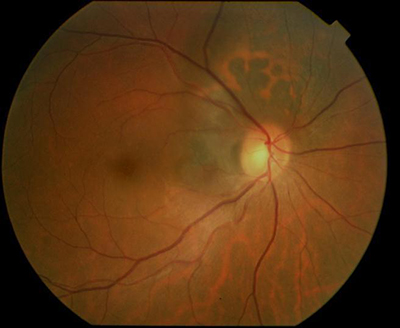 Choroidal melanoma can grow near, touch and even cover the optic nerve. Like other choroidal melanomas, they can exhibit orange pigment on its surface, subretinal fluid (localized retinal detachment), and thickness. The choroidal melanoma in the next photograph exhibits all three findings.
Choroidal melanoma can grow near, touch and even cover the optic nerve. Like other choroidal melanomas, they can exhibit orange pigment on its surface, subretinal fluid (localized retinal detachment), and thickness. The choroidal melanoma in the next photograph exhibits all three findings.