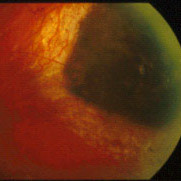
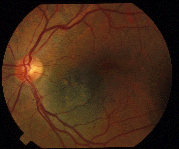
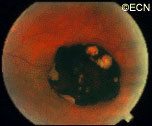
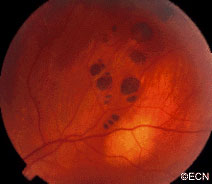
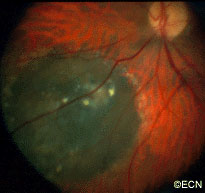
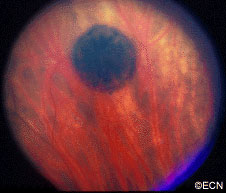
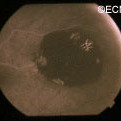
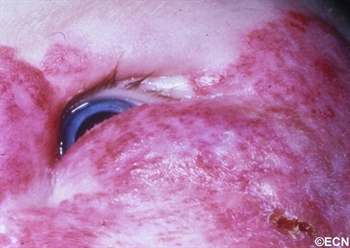
Eye Plaque Results: Treatment of Choroidal Melanoma
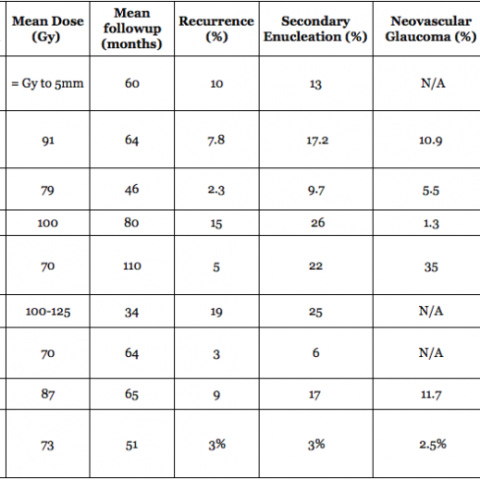
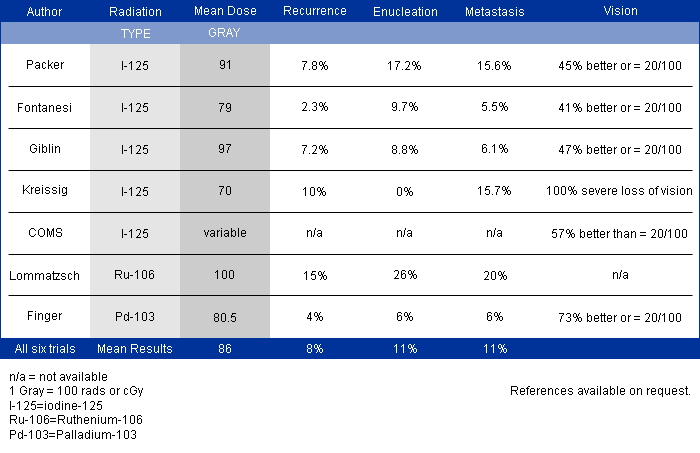
In a (2009) publication*, Dr. Finger created a table summarizing the results of the largest major clinical studies of radiation for choroidal melanoma at approximately 5 years follow up.
Chart: Comparison of Results after Plaque Radiation Therapy
(Click to Enlarge and View the Full Chart)
Overview Reference
Radiation Therapy for Choroidal Melanoma. Finger PT.
Survey of Ophthalmology (Review Article) 42:215-32, 1997
Palladium-103 ophthalmic plaque radiation therapy for choroidal melanoma: 400 treated patients. Finger PT, Chin KJ, Duvall G for The Palladium-103 for Choroidal Melanoma Study Group. Ophthalmology 2009;116:790-6.
References
Melia BM, Abramson DH, Albert DM, et al. Collaborative ocular melanoma study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years. COMS report No. 16. Ophthalmology. 2001;108:348-366.
Jampol LM, Moy, CS, Murray TG, et al. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS report no. 19. Ophthalmology. 2002 Dec;109(12):2197-206.
Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol. 2006 Dec;124(12):1684-93.
Packer S, Stoller S, Lesser ML, et al. Long-term results of iodine 125 irradiation of uveal melanoma. Ophthalmology. 1992;99:767-774.
Fontanesi J, Meyer D, Xu S, et al. Treatment of choroidal melanoma with I-125 plaque. Int J Radiat Oncol Biol Phys. 1993;26:619-623.
Lommatzsch PK. B-irradiation of choroidal melanoma with 106-Ru/106-Rh applicators: 16 years’ experience. Arch Ophthalmol. 1983;101:713-717.
Gragoudas ES, Seddon JM, Egan K, et al. Long-term results of proton beam irradiated uveal melanomas. Ophthalmology. 1987 Apr;94(4):349-53.
Char DH, Kroll SM, Castro J. Ten-year follow up of helium ion therapy for uveal melanoma. Am J Ophthalmology 1988:125:81-9.
Brokvina AF, Zarubei GD. Ciliochoroidal melanomas treated with a narrow medical proton beam. Arch Ophthalmol 1986;104:402-4.
Finger PT, Chin KJ, Duvall BS for The Palladium-103 for Choroidal Melanoma Study Group. Palladium-103 Ophthalmic Plaque Radiation Therapy for Choroidal Melanoma: 400 Treated Patients. Ophthalmology 2009;116:790-6.
A gold eye-plaque (front and back) and silicone insert – seed carrier.
Ophthalmic plaque radiation therapy is the most commonly used “eye and vision-sparing” treatment for choroidal melanoma (around the world). A radioactive plaque is a small, dish-shaped device that contains a radioactive source.
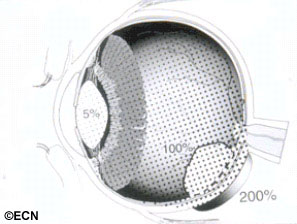
In order to visualize the intraocular tumor during surgery, a bright light is placed on the eye. This is called transillumination. The thick and pigmented tumor within the eye blocks the light, creating a shadow or dark spot on the wall of the eye.
In order to visualize the intraocular tumor, during surgery, a bright light is placed on the eye. The thick tumor within the eye commonly blocks the light creating a shadow or dark spot on the wall of the eye.
The edges of the shadow is marked (on the sclera) with a temporary tissue marker.Low-energy radioactive eye-plaques contain rice-sized iodine-125 or palladium-103 seeds that emit low energy photon (23-28 KeV) radiation. This radiation is effectively blocked by the gold-backing of the plaque creating a directional source.
Typically, the radioactive eye-plaque is then sewn onto the eye as to cover the intraocular tumor shadow, plus a 2-3 mm “free-margin.” This extra margin is used to make sure that the tumor is always under the plaque during radiation. With the plaque in place, radiation is continuously delivered over 5 to 7 days, and then removed.
The first plaques incorporated the radioactive material Cobalt-60 and later included Ruthenium-106 and Iodine-125. In 1990, palladium-103 (Pd-103) became available for the first time for treatment of intraocular tumors.
Typically, the radioactive eye-plaque is then sewn onto the eye as to cover the intraocular tumor shadow, plus a 2-3 mm “free-margin.”
Multiple dose comparisons (versus iodine-125) showed that palladium-103 (Theragenics Corp. Buford, Georgia) offered a more favorable intraocular dose distribution in most cases. It delivered slightly more irradiation to the tumor while allowing less to be absorbed by the patients surrounding normal structures. Less radiation also reaches the operating surgeon and health-care personnel. Dr. Finger recommends the use of palladium-103 for ALMOST ALL of his patients with choroidal melanoma.
Long-term results on plaque-treated patients are now available.
Reported Results after Ophthalmic Plaque Radiation Therapy
Related Links
The American Brachytherapy Society consensus guidelines for plaque brachytherapy of uveal melanoma and retinoblastoma – Download the PDF
Dosimetry of 125I and 103Pd COMS eye plaques for intraocular tumors: Report of Task Group – Download the PDF
In ophthalmology we are blessed with many well-defined clinical indications where the potential benefits outweigh the risks of treatment; where we clearly help our patients. This is not always the case in treatment of “small choroidal melanomas.” [1-3] At first blush, the choice appears simple, loss of vision (in one eye) versus death (due to metastatic melanoma). A closer look reveals controversy’s in the diagnostic criteria used for small choroidal melanomas, that treatment may or may not risk loss of central vision and that observed tumor-growth may be the best predictor of malignancy.
Diagnosing Small Choroidal Melanomas
In the past, eyes were enucleated with the diagnosis of choroidal melanoma only to find simulating or benign lesions on histopathology.[4] The advent and evolution of modern diagnostic techniques such as indirect ophthalmoscopy, ultrasonography, photography, angiography and subspecialty centers have improved our diagnostic ability to detect small changes in tumor size and patterns of leakage.
Tumor-specific characteristics have been found to carry diagnostic value. [5][6] For example, a choroidal tumor with orange pigment on its surface, thickness of greater than 2 mm and subretinal fluid is very likely to be a small malignant choroidal melanoma. Mainly using these criteria, The Collaborative Ocular Melanoma Study (COMS) physicians were reported to correctly diagnose medium and large-sized choroidal melanomas in 99.6% of cases.[7] Documented tumor growth [over a relatively short period of time (typically months)] is commonly used to differentiate a suspicious choroidal nevus from a small choroidal melanoma.[8] Observation for growth is used because there currently exists no safe and effective method to obtain a biopsy (cytopathologic proof) of tumors that small (less than 10 x 10 wide x 3 mm thick).
The Case for “Observation as Treatment”
Many eye cancer specialists watch small choroidal melanomas for evidence of tumor-growth prior to treatment. This is because current belief is that “small choroidal melanomas” carry a low risk (6-10%) for metastases and that all current treatments risk severe vision loss.[9-11] Current observation of small choroidal melanomas is justified by the concept that “tumor-growth demonstrates malignancy.” In practice, documented tumor growth both reassures the doctor that the tumor is malignant and reassures the patient that treatment is indicated. Specifically, the risk of treatment-related loss of vision is more than offset by a reduction in the probability of metastasis and tumor-induced vision loss.
This is particularly true for patients with small choroidal melanomas close to the fovea, monocular or systemically ill patients. In these cases, serial observation may allow for years of useful vision prior to treatment. For all patients, the case for observation of small melanoma growth has been governed by the potential benefit of vision preservation (in the affected eye).
However, the risk of vision loss in radiation treated eyes has recently diminished. In 2006, Finger discovered and others have found that anti-VEGF medications can preserve vision in eyes affected by radiation maculopathy and optic neuropathy. These studies suggest that radiation damage may no longer inevitably lead to vision loss and changes the balance between treatment and observation.
The Case for Immediate Treatment
Once an eye cancer specialist is convinced that a tumor is a malignant (albeit small) choroidal melanoma, treatment becomes the most reasonable choice. In support of this approach, one can cite Packard’s and the Collaborative Ocular Melanoma Study’s (COMS) findings that increased tumor size (specifically largest tumor diameter – LTD) was associated with an increased risk of metastatic death.[10,11] Therefore, it is reasonable to assume that waiting for documentation of malignant melanoma growth increases (albeit marginally) a patient’s risk for metastases.
This is no surprise. Increasing tumor size had been associated with an increased incidence of metastases in cancers of the eye, breast, lung, and colon, and cutaneous melanoma. Throughout medical oncology there exists a fundamental understanding that early treatment of cancer saves lives. This concept has led to the development of national strategies aimed at early cancer detection.
I could find no cancer monitoring or treatment programs that promote observation of cancer growth when treatment is available. Similarly, ophthalmic oncologists would not promote observation of small malignant eyelid or conjunctival melanomas.
Currently, all patients with small choroidal melanoma can be offered eye and vision-sparing treatments. Particularly, if one uses the tumor’s apex as the prescription point for plaque radiation therapy (as recommended by the American Brachytherapy Society), small choroidal melanomas require particularly small treatment volumes that should be associated with less resulting vision loss.[12]
Recent evidence suggests that should radiation retinopathy and optic neuropathy occur, it is treatable with anti-VEGF agents (eg. Avastin or Lucentis).13 Lastly, immediate treatment will prevent the tumor enlargement associated increased risk of metastatic melanoma.
Continued Observation Once Definitive Growth Has Been Documented
Very few eye cancer specialists would recommend continued observation once definitive tumor-growth has been documented. Choroidal nevi grow, but typically exhibit small changes over years of observation. Relatively rapid (e.g. months) and measurable tumor growth is consistent with malignancy. Therefore as currently practiced widely, documented rapid tumor enlargement indicates that a “suspicious choroidal nevus” is actually a malignant choroidal melanoma, and will (by continued enlargement and/or secondary retinal detachment) cause loss of vision. Once a small melanoma has grown, treatment offers the best chance for preservation of both life and vision in the affected eye.
Hope from the Future
Current research offers the potential to aid in the diagnosis of small choroidal melanoma. Onken, Harbour et al are exploring the use of molecular and genetic markers to assess a tumors metastatic potential.14 Genetic studies suggest that monosomy 3, together with other genetic markers may define a tumor’s metastatic potential. Similarly, physiologic-radiographic imaging (such as PET/CT) may offer the potential to assess a tumors metabolic activity and metastatic potential.15-18
Expect new methods to address treatment-related complications, decrease morbidity and preserve functional vision.19 Lastly, we will see earlier detection of metastatic disease.16,17 When metastatic disease is found, local therapies will be largely abandoned in favor of systemic treatment.
Doing the Least Harm
This editorial explores the controversy’s surrounding treatment of small choroidal melanomas. “Observation as treatment” for suspected small choroidal melanomas offers the patient time (without the risk of treatment-related vision loss) at the risk (small increase in the probability) of death from metastatic choroidal melanoma. This is why, despite our advances in the diagnosis and treatment of these tumors, there exists considerable controversy “among experts” about what characteristics differentiate small choroidal melanomas from indeterminate choroidal tumors and when treatment is warranted.2,8,20
Current practice dictates that eye cancer specialists continue to perform clinical assessments, classify small choroidal tumors and discuss the potential risks and benefits of observation, biopsy and treatment with each patient. Keep in mind that your doctors recommendation will be influenced by his or her experience with treatment-related risk for vision loss versus that due to metastatic melanoma. Further, physicians will determine the patient’s ability to understand what has been presented and recommend an approach to do the “least” harm.
Summary
Until better methods of differentiation are available, “Observation as treatment” will continue to be the standard of care for benign and suspicious choroidal nevi, as well as most small indeterminate choroidal tumors. Treatment will be recommended for small malignant choroidal melanomas, particularly if those tumors are documented to grow.
References
Barr CC, Sipperley JO, Nicholson DH. Small melanomas of the choroid. Arch Ophthalmol 1978;96(9):1580-2.
Char DH. The management of small choroidal melanomas. Surv Ophthalmol 1978;22(6):377-86.
The Collaborative Ocular Melanoma Study Group. Mortality in patients with small choroidal melanoma. COMS report no. 4. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997;115(7):886-93.
Chang M, Zimmerman LE, McLean I. The persisting pseudomelanoma problem. Arch Ophthalmol 1984;102(5):726-7.
Augsburger JJ, Schroeder RP, Territo C, et al. Clinical parameters predictive of enlargement of melanocytic choroidal lesions. Br J Ophthalmol 1989;73(11): 911-7.
The Collaborative Ocular Melanoma Study Group. Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group.” Arch Ophthalmol 1997;115(12): 1537-44.
Collaborative Ocular Melanoma Study Group: Accuracy of diagnosis of choroidal melanomas in the Collaborative Ocular Melanoma Study. COMS Report No. 1. Arch Ophthalmol 108:1268-1273, 1990.
Augsburger JJ. Is observation really appropriate for small choroidal melanomas. Trans Am Ophthalmol Soc 1993;91:147-68; discussion 169-75.
Finger PT. “Radiation therapy for choroidal melanoma.” Surv Ophthalmol 1997;42(3):215-32.
Packard R B. “Pattern of mortality in choroidal malignant melanoma.” Br J Ophthalmol 1980;64(8):565-75.
The Collaborative Ocular Melanoma Study Group. Mortality in patients with small choroidal melanoma. COMS report no. 4. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997;115(7):886-93.
Nag, S, Quivey JM, Earle JD, Followill D, Fontanesi J, Finger PT. The American Brachytherapy Society recommendations for brachytherapy of uveal melanomas. Int J Radiat Oncol Biol Phys 2003;56(2):544-55.
Finger PT. Radiation retinopathy is treatable with anti-vascular endothelial growth factor bevacizumab (Avastin). Int J Radiat Oncol Biol Phys 2008;70:974-7.
Onken MD, Worley, LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res 2004;64(20):7205-9.
Reddy S, Kurli M, Tena L, Finger PT Whole body PET/CT imaging: Detection of Choroidal Melanoma. Br J Ophthalmol 2005;89(10):1265-9.
Finger PT, Kurli M, Reddy S, Tena LB, Pavlick AC (2005) Whole body PET/CT for initial staging of choroidal melanoma. Br J Ophthalmol 2005;89(10):1270-4.
Finger PT, Kurli M (2005) Laser photocoagulation for radiation retinopathy after ophthalmic plaque radiation therapy. Br J Ophthalmol 89;730-8.
18-Fluorine-labelled 2-deoxy-2-fluoro-D-glucose positron emission tomography/computed tomography standardised uptake values: a non-invasive biomarker for the risk of metastasis from choroidal melanoma. Br. J. Ophthalmol 2006;90(10):1263-6.
Kurli M, Reddy S, Tena LB, Pavlick AC, Finger PT (2005) Whole body positron emission tomography / computed tomography (PET/CT) staging of metastastic choroidal melanoma. Am J Ophthalmol 2005;140(2):193-9.
Murray TG (1997). Small choroidal melanoma. Arch Ophthalmol 115(12): 1577-8.
Related Links
WOC: Bio-informatics Can Solve the Mystery of Treatment of Small Choroidal Melanomas – Download the PDF Here
This article appeared in the November / December, 2012 issue of Retina Today
By Paul T. Finger, MD, FACS
During the 2012 American Academy of Ophthalmology’s (AAO) Retina Subspecialty Day, there was a panel, “Tumor Management: Radiation, Retinopathy and Masquerade,”moderated by Evangelos Gragoudas, MD. I was a panelistalong with Jerry Shields, MD; David Abramson, MD; Brenda Gallie, MD; and Arun Singh, MD. A case of radiation optic neuropathy (RON) with radiation maculopathy(RM) was shown, and I was asked my opinion concerning management. As is my practice, I responded that I typically wait for the first signs of a patient’s radiation vasculopathy (eg, edema, exudate, cotton wool spots, hemorrhage) to discuss the use of anti-VEGF intervention. As part of informed consent, I tell them that few patients escape an episode of untreated RON or RM with useful vision. Although we all have seen selected cases of spontaneous resolution of radiation vasculopathy and “burnout” over time, the final vision outcomes are typically poor.1-3
In 1997, I first published cases in which intravitreal anti- VEGF injections were found to successfully decrease vascular transudation associated with RM and RON.4,5 Since that time we have published several clinical case series showing similar findings with vision preservation.6-9 Now there are many worldwide reports that radiation-induced macular retinal vessel transudation can be controlled with periodic intravitreal anti-VEGF medication.10-13 Unfortunately, at the AAO I also learned that in some countries only 2 to 3 monthly anti-VEGF injections are approved for each patient. Even in the United States some patients suffer from living “just too far” away for compliance. This is analogous to giving just 1 or 2 shots of insulin for treatment of a lifetime of diabetes. Such approaches are inadequate and doomed to failure. It is my opinion that most intraocular radiation vasculopathy will be successfully suppressed only with consistent periodic intravitreal anti-VEGF therapy.
When Should We Stop Therapy?
Unfortunately, we have witnessed a few cases in which patients significantly delayed their follow-up anti-VEGF treatment and developed off-treatment recurrent optic disc and/or macular edema. Though these cases typically respond a second time after restarting intravitreal anti-VEGF therapy, measurable damage occurred in the interim. These cases cement my conviction concerning the efficacy and need for continuous anti-VEGF treatment for radiation vasculopathy. As a result of long-term experience, I typically counsel patients that periodic intravitreal anti-VEGF treatment for intraocular radiation vasculopathy is analogous to insulin treatment for diabetes. It merely suppresses a progressive disease. The more consistent we are with treatment, the more likely vision will be preserved.
What Anti-VEGF Dose Is Best?
Anti-VEGF strength also appears to make a difference. Analyses of our pilot data was presented at the 2012 American Society of Retinal Specialists and Retina Society meetings. In the Genentech and investigator-sponsored study, 2.0 mg ranibizumab was used to treat cases that were not suppressed by standard dose therapy.14 This series included both post-plaque and post-external beam radiation therapy (EBRT) patients. We found that in patients who were not responding to standard-dose anti-VEGF therapy, their radiation maculopathy responded to 2.0 mg ranibizumab. Because more anti-VEGF worked better, there must be more VEGF in these irradiated eyes.
Why might there be more VEGF in these eyes?
First, choroidal melanomas have been noted to produce VEGF. Second, radiation is known to induce a progressive obliterative vasculopathy with secondary VEGF-producing ischemia. Therefore, it is reasonable to assume that, compared with exudative macular degeneration, eyes irradiated for cancer contain higher levels of VEGF. This includes both those with intraocular tumors and eyes treated with
EBRT for metastatic, orbital and sinus tumors.
What does all this teach us?
Published studies that show radiation dose to the macula can be used to predict the incidence of radiation maculopathy. Therefore, it is reasonable to assume there is a need to decrease the radiation dose used to treat tumors in the eye.3,15,16 Decreased radiation dose (and dose rates) will lead to less, or more treatable, intraocular radiation vasculopathy.
How Can We Decrease the Radiation Dose?
Several of my ophthalmic oncology colleagues do not share my experiences and views about anti-VEGF therapy. Therefore, I have tried to look for an objective reason why their results might differ. Basic radiation therapy teachings tell us that the higher the dose and the faster the dose rate the greater the complication rates. So let us compare commonly used radiation treatments and make some simple recommendations.
Ruthenium-106 Plaque Therapy
In Europe, the most commonly used plaques contain ruthenium-106 (Ru-106). Ru-106 plaques emit beta irradiation, particles that typically travel 5 to 6 mm into the eye and have a very high base-to-apex dose gradient. For example, in treatment of a 5-mm high tumor, the base (sclera, choroid, and retina) dose can be 3 to 4 times that seen using an equivalent apex dose of iodine-125 (I-125).17 In addition, when a Ru-106 plaque is placed near the macula, that macular retina will receive much more and faster irradiation compared with either I-125 or palladium-103 (Pd-103) seeded plaques. Because dose and dose-rate matter, the higher Ru-106 radiation dose will produce a more severe retinopathy or optic neuropathy that is less likely to respond to anti-VEGF medications.
External Beam Radiation Therapy
Proton beam therapy doses are typically numerically similar to prescription doses used to the choroidal melanoma apex during I-125 and Pd-103 plaque therapy. However, these 2 treatments are actually like radiation apples and oranges. Consider that plaque brachytherapy is delivered slowly (typically over 5 to 7 days), whereas protons are given in several daily or alternate-day sessions of less than 15 minutes each. These high-dose-rate sessions are not radiobiologically equivalent to slow-dose brachytherapy. It is generally accepted that the faster you give a radiation dose the more the long-term side effects. Treatment plans should reflect a balance between a dose fast enough to kill the tumor but slow enough to preserve normal tissues. This also applies to standard photon-based EBRT for choroidal metastasis, orbital and sinus tumors. At The New York Eye Cancer Center we currently recommend EBRT dose rates of 180-200 cGy per day as well as treating to the lower range of acceptable total treatment doses.3 In this way, we are preparing the patient to avoid or delay intraocular radiation vasculopathy or make it less severe and more treatable. An analogy can be taken from the heat radiation (flame) of a candle. If you take the heat energy all at once (high-dose rate) by putting your finger on the flame and leaving it there, you will burn the skin on your finger. But if you absorb the same amount of heat energy over time (by moving your finger back and forth through the flame for a longer time) it will just become warmed. The former burn is more difficult to treat. Similarly, high-dose-rate radiation is less likely to respond to periodic intravitreal anti-VEGF therapy.
I-125 and Pd-103 Plaque Therapy
In the United States, most eye cancer specialists use low-energy I-125 or Pd-103 plaque therapy.3,18 However, I have found that low-energy plaque therapy is delivered in several ways. Though the first series and the Collaborative Ocular Melanoma Study suggested a 5 to 7 day continuous treatment, some centers are decreasing the course of treatment to 3 days (a higher dose rate). As mentioned, this shorter treatment interval should increase late normal tissue side effects. Another problem is a legacy of Collaborative Ocular Melanoma Study (COMS) dose planning. Here, the COMS
required a minimum dose of 85 Gy to 5 intraocular millimeters. That is, for a 2.5 mm high tumor one was required to treat to a minimum 5 mm height. Thus, the COMS dose plan increased the dose and dose rate to those smaller tumors while increasing the dose (and dose rate) to normal ocular structures (eg, the fovea and optic nerve). It is no wonder that the COMS reported the worst visual acuity results of any plaque study. It is reasonable to assume that any center using that legacy COMS dosing strategy will increase both the incidence and severity of RM and optic neuropathy as well as hindering patient response to anti-VEGF therapy. In 2003, the American Brachytherapy Society eliminated the minimum 5 mm COMS requirement.19
Our Current Strategies
At The New York Eye Cancer Center we perform presurgical mathematical modeling to compare the intraocular dose distribution of either I-125 or Pd-103 plaque therapy for each patient. For an equivalent tumor dose; we choose the source that will relatively spare the macula, optic nerve, or, if suprathreshold, the lowest organ dose. In comparison to I-125, most patients are better off with the Pd-103 radionuclide.18,20 We do not use Ru-106 plaques for several reasons beyond the scope of this article. Similarly, for our standard EBRT patients, we discuss our concerns with their treating radiation oncologist. We urge a limit of 180 to 200 cGy daily dose rates and the lowest acceptable total doses. We explain that the eye is relatively sensitive to irradiation. In treatment of selected posterior choroidal melanomas that are at high risk for RM or RON, I first offer laser demarcation to suppress the ischemic drive likely contributing to elevated intraocular VEGF.21 Laser delivered in 1 or 2 sessions may avoid the need for periodic intravitreal injections. However, in cases where laser does not work, is not possible, or when the tumor is beneath the fovea, anti-VEGF therapy offers the patient the best chance for vision preservation. We continue to treat patients with anti-VEGF agents for RM and RON related to treatment of ocular lymphoma, uveal metastasis, and cancers of the lacrimal gland, sinus, and ocular adnexa. Regardless of the source, our current strategy is initial monthly dosing of bevacizumab (1.25 mg/0.05 mL) or ranibizumab (0.5 mg) to determine the degree of the clinical response. Encouraged by early and then persistent suppression of macular edema, hemorrhages, and exudates, we titrate the number of injections (to 6-, 8-, and rarely 12-week intervals) needed to stabilize patients over time. Since 1995, there has been only 1 case in which we have been able to completely discontinue maintenance dosing. In recalcitrant cases, the level of VEGF may be too high for standard intravitreal anti-VEGF agents to overcome. These patients may benefit from a higher dose. Bevacizumab is currently available in higher 2.0 and
2.5 mg doses. However, the increased drug volume has increased transient post-injection vision loss and secondary increased intraocular pressures. We recognize that radiation retinopathy is a progressive disease and that dose escalation strategies may offer our patients additional time to forestall vision loss. To date, intravitreal anti-VEGF therapy has allowed us to preserve useful vision in most of our patients over the past 6 years.
Paul T. Finger, MD is the Director of The New York Eye Cancer Center and of Ocular Tumor Services at The New York Eye and Ear Infirmary, New York University School of Medicine, and affiliated hospitals. He states that his research is supported by The Eye Cancer Foundation and that he holds a US patent titled “Anti-VEGF Treatment for Radiation Induced Vasculopathy,” US Patent 7553486, and a trademark “Think of sunglasses as sunblock for your eyes,” US Trademark 75-779211.
Finger PT. Radiation therapy for orbital tumors: concepts, current use and ophthalmic radiation side effects. Surv Ophthalmol. 2009;54(9):545-568.
Finger PT. Anti-VEGF bevacizumab (Avastin®) for radiation optic neuropathy. Am J Ophthalmol. 2007;143:335-338.
Finger PT, Chin K. Anti–vascular endothelial growth factor bevacizumab (Avastin) for radiation retinopathy. Arch Ophthalmol. 2007;125:751-756.
Finger PT. Radiation retinopathy is treatable with anti–vascular endothelial growth factor bevacizumab (Avastin). Int J Radiat Oncol Biol Phys. 2008;70:974-977.
Finger PT, Mukkamala SK. Intravitreal anti-VEGF bevacizumab (Avastin) for external beam related radiation retinopathy. Eur J Ophthalmol. 2011;21:446-451.
Finger PT, Chin J. Antivascular endothelial growth factor bevacizumab for radiation optic neuropathy: Secondary to plaque radiation therapy. Int J Radiat Oncol Biol Phys. 2012;82:789-798.
Solano JM, Bakri SJ, Pulido JS. Regression of radiation-induced macular edema after systemic bevacizumab. Can J Ophthalmol. 2007;42(5):748-749.
Mason JO 3rd, Albert MA Jr, Persaud TO, Vail RS. Intravitreal bevacizumab treatment for radiation macular edema after plaque radiotherapy for choroidal melanoma. Retina. 2007;27(7):903-907.
Gupta A, Muecke JS. Treatment of radiation maculopathy with intravitreal injection of bevacizumab (Avastin). Retina. 2008;28(7):964-968.
Arriola-Villalobos P, Donate-Lopez j, Calvo-Gonzalez C, Reche-Frutos J, Alejandre-Alba n, Diaz-Valle D. Intravitreal bevacizumab (Avastin) for radiation retinopathy neovascularization. Acta Ophthalmol. 2008;86(1):115-116.
Finger PT. Clinical response to intravitreal high dose (2.0) ranibizumab for radiation maculopathy. Paper presented at: the 2012 Retina Society meeting; October 4-7, 2012; Washington, DC.
Finger PT, Chin KJ, Yu GP; for the Palladium-103 for Choroidal Melanoma Study Group Risk factors for radiation maculopathy after ophthalmic plaque radiation for choroidal melanoma. Am J Ophthalmol. 2010;149(4):608-615.
Khan N, Khan MK, Bena J, Macklis R, Singh AS. Plaque brachytherapy for uveal melanoma: a vision prognostication model. Int J Radiat Oncol Biol Phys. 2012;84:e250-290.
Finger PT. Radiation therapy for choroidal melanoma. Surv Ophthalmol. 1997;42:215-232.
Finger PT, Chin KJ, Duvall G, for The Palladium-103 for Choroidal Melanoma Study Group Palladium-103 ophthalmic plaque radiation therapy for choroidal melanoma: 400 treated patients. Ophthalmology. 2009;116:790-796.
Nag S, Quivey JM, Earle JD, Followill D, Fontanesi J, Finger PT, for the American Brachytherapy Society. The American Brachytherapy Society recommendations for brachytherapy of uveal melanomas. Int J Radiat Oncol Biol Phys. 2003;56(2):544-555.
Chiu-Tsao ST, Astrahan MA, Finger PT, et al. Dosimetry of (125)I and (103)Pd COMS eye plaques for intraocular tumors: Report of Task Group-129 by the AAPM and ABS. Med Phys. 2012;39(10):6161-6184.
Finger PT, Kurli M. Laser photocoagulation for radiation retinopathy after ophthalmic plaque radiation therapy. Br J Ophthalmol. 2005;89:730-738.
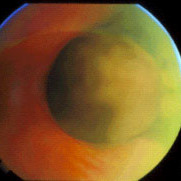
Note the flat, black well circumscribed lesion with areas of retinal pigment epithelial atrophy.
The retinal pigment epithelium (RPE) is a pigmented layer of the retina which can be thicker than normal at birth (congenital) or may thicken later in life. Areas of retinal pigment epithelial (RPE) hypertrophy usually do not cause symptoms. They are typically found during routine eye examinations.
Congenital retinal pigment epithelial hypertrophy (CHRPE) is usually found before patients reach 30 years of age. They may enlarge with time, but are not malignant. CHPRE has been an association with Gardner’s Syndrome (familial colonic polyposis).
This is a case of congenital hypertrophy of the retinal pigment epithelium, “bear-tracks.”
Therefore, if your eye care specialist has told you that you have CHPRE( pronounced CHER PEE), it is reasonable to tell your primary care physician or gastroenterologist (if you have one) so that he or she may recommend the frequency of colon screening tests.
In contrast, acquired retinal pigment epithelial hypertrophy (RPEH) is typically found later in life. They are typically jet-black to gray, flat, with a halo around its edges. Variable in size, RPEH lesions may develop lacunae of lightly colored areas of atrophy (see image above). These
This area of retinal pigment epithelial hypertrophy demonstrates a blue hue.
tumors are more commonly found in the peripheral retina where thickness is more difficult to judge by ophthalmoscopy.
Symptoms
Almost all patients with retinal pigment epithelial hypertrophy do not have symptoms. These pigmented intraocular lesions are found by eye care specialists during dilated examination of the inside of the eye (ophthalmoscopy). Eye tumor specialists can typically differentiate between retinal pigment epithelial hypertrophy and melanoma by clinical examination (without a biopsy).
Diagnosis
A small area of retinal pigment epithelial hypertrophy. It appears well circumscribed and with areas of relative lucency at the edges.
Retinal pigment epithelial hypertrophy (CHRPE and RPEH) can be diagnosed by ophthalmic examination. The eye examination will concentrate on the appearance of the retinal pigment epithelial hypertrophy. RPEH lesions tend to be black or atrophic. They may be surrounded by a halo of less pigmented tissue or exhibit a sharp demarcation line.
Ultrasonography typically shows that RPE hypertrophy is flat to minimally elevated and slightly hyper-reflective.
Fluorescein angiography of RPE hypertrophy typically demonstrates blockage of fluorescence (except in the areas of atrophy which are hyperfluorescent).
Optical coherence tomography (OCT) of RPE hypertrophy will demonstrate both thickening and thinning. The overlying retina is thinned, the retinal pigment epithelium is both thickened or can be thinned. The underlying choroid is typically thinned.
Treatment
Photographic documentation of these lesions is recommended for future comparison. Ultrasonography and fluorescein angiography is typically used to differentiate RPE hypertrophy from uveal melanoma and certain rare intraocular tumors. Serial observation is warranted in that RPE hypertrophy can enlarge over time.
A large periocular hemangioma involves both the upper and lower eyelids. The eye is able to open but there is an astigmatism induced by a mass effect from the lower eye lid.
Children can either be born with or develop reddish “strawberry” colored tumors on or around their eyes. This is one of the most common tumors of infancy. It is 3 times as frequent in girls and can run in families.
Symptoms
Periocular hemangioma of childhood can be large, and commonly grow during the first year of life, but also tend to get smaller (involute) over the following 2 years.
Periocular hemangioma of childhood can extend into the orbit(behind the eye) and push the eye forward (proptosis), make the eyes misaligned (strabismus), or can cause the eyelid to droop (ptosis).
Children with periocular hemangioma of childhood can have hemangiomas in other parts of their bodies, so a pediatric consultation is necessary. If the hemangiomas are multiple or on the jaw or neck, a pulmonary consultation is necessary to rule out upper respiratory tract involvement. Consider the PHACES syndrome (anomalies of the Posterior fossa, Hemangiomas, the Arteries, Cardiac, Eye, Sternum) which is more commonly seen in girls.
Diagnosis
Hemangioma can be diagnosed by ocular examination with magnetic resonance imaging (MRI). Rarely, a small biopsy may be required to confirm the clinical diagnosis.
Treatments
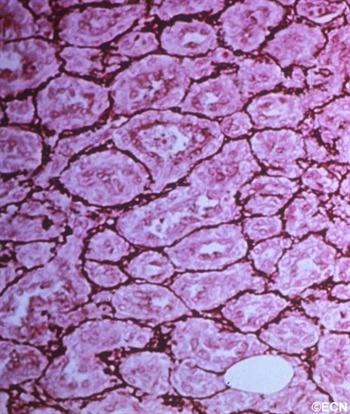
Histopathology reveals multiple well differentiated vascular channels. No atypical cells are seen.
Since periocular hemangioma of childhood is a benign tumor (not a cancer), immediate treatment is often not necessary. In fact, after an initial growth phase, many of these tumors will get smaller by themselves. Most patients can be followed for evidence of spontaneous remission.
Treatment is urgently indicated if the periocular hemangioma of childhood is found to harm the proper development of vision in the affected eye in infants and young children (amblyopia), and for psychosocial reasons in older children and adults.
Let me explain Amblyopia: In order for an eye to achieve its best possible potential for vision, two things are necessary. First, proper images must be focused on the retina and second, the brain must receive those images. During the first 10 years of life, there is a process where images are collected and the brain learns to understand those images. If an eye is blocked by the tumor (or the eye lid), or if the eyes are not aligned, or if the eye is not able to focus images on the retina, the child’s eye-brain connection will not develop. That is, the child will not learn to see from that eye (a problem called amblyopia). Urgent treatment of periocular hemangioma of childhood can be necessary be to prevent amblyopia.
Periocular hemangioma of childhood has been treated with surgery, laser-surgery, radiation, and drugs (intralesional steroids and systemic beta-blockers). When possible, treatment of periocular hemangiomas of childhood involves injections of steroid into the tumor. In comparison to taking the medicine by mouth (PO) or by vein (IV), this approach has the advantage of putting the medicine right into the tumor. An acute effect, but does carry risk of tumor and orbital hemorrhage.
References
Haik B, Karcioglu Z, Gordon RA, Pechous BP. Capillary hemangioma (infantile periocular hemangioma). Survey of Ophthalmology 1994;38:399-426.
Kushner BJ. Hemangiomas. Archives of Ophthalmology 2001;118;835-836.
Rhabdomyosarcoma is the most common primary malignancy of the orbit in children. It can also occur in adults, though the average age of patients affected by rhabdomyosarcoma is 7 – 8 years.
Symptoms
Most parents first notice a droopy eyelid (called ptosis), and that the eye is more prominent (called proptosis), or that their child has a tumor under the conjunctival membrane that covers the eye (globe). Rhabdomyosarcoma is usually found in the superonasal orbit (that is under the upper lid near the nose).
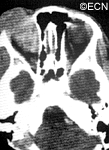
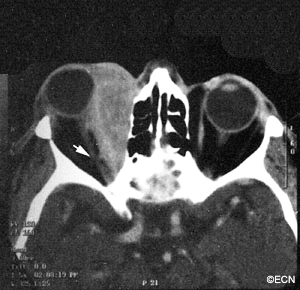
CT of Orbital Rhabdomyosarcoma
Diagnosis
Computed axial tomography (CT-scan) and magnetic resonance imaging (MRI) typically show a mass adjacent to or attached to one of the ocular or orbital muscles. CT is particularly helpful because it offers the best evidence if the orbital bones have been invaded by the rhabdomyosarcoma tumor.
Treatments
Rhabdomyosarcoma can grow rapidly and if the tumor grows into the brain or spreads to the lung, survival is poor. Prompt biopsy of a rhabdomyosarcoma followed by a combination of chemotherapy and irradiation offers the best chance of survival. In fact, recent reports suggest that current treatments offer greater than 90% survival from rhabdomyosarcoma.
Patients will develop problems typically seen after chemotherapy and irradiation of the eye, but if there is no recurrence after 3 years, it is likely that the rhabdomyosarcoma has been controlled.
By local growth sclerosing orbital pseudotumors can cause bulging of the eye (proptosis).
Sclerosing orbital pseudotumor is uncommon. Due to unknown reasons, these tumor behave differently than other types of pseudotumor of the orbit. They grow more slowly, cause less pain, and are characterized by scarring (hardening of the tumor tissue).
Symptoms
Sclerosing orbital pseudotumor is not cancer. But, by local growth it can cause bulging of the eye (proptosis), double vision (diplopia) and loss of vision. Sclerosing orbital pseudotumor can (rarely) extend into the sinuses, brain, and other orbit.
Diagnosis
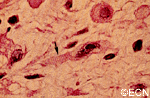
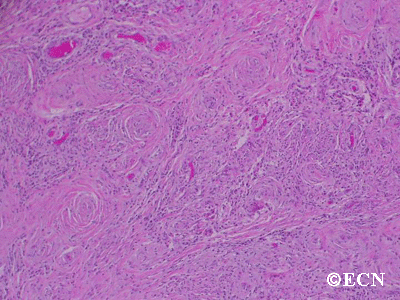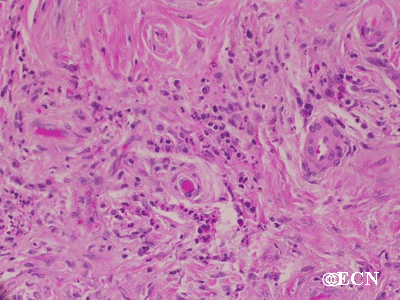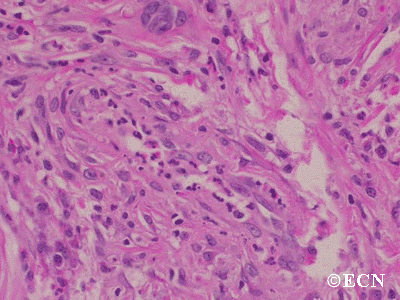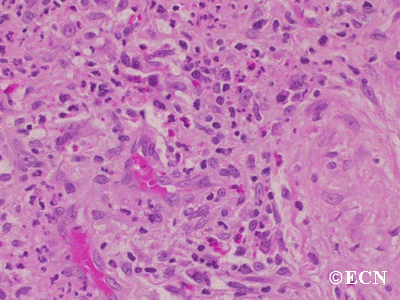
Histopathology reveals large amounts of fibrous tissue.
Sclerosing orbital pseudotumor is usually diagnosed by biospy (orbitotomy).
Once the diagnosis is confirmed by pathology, systemic testing to rule out specific infectious and inflammatory causes should be performed. For example, an ANCA blood test and a chest x-ray should be performed to rule out Wegener’s Granulomatosis. Many of these patients have a past medical history of sinusitis, sinus surgery, or inhalation drug abuse. Therefore, concurrent treatable sinus disease should be addressed.
Treatments
In this case, computed tomographic (CT) scanning demonstrates a mass in the nasal orbit. The arrow demonstrates the optic nerve on stretch. The eye wall (sclera) is indented and the eye pushed out (proptosis).
Sclerosing orbital pseudotumors are less responsive to steroid therapy. Most cases are treated with combinations of surgery, steroid therapy, radiation and chemotherapy depending upon the clinical picture and the patient’s response to treatment.
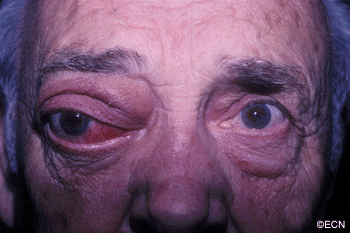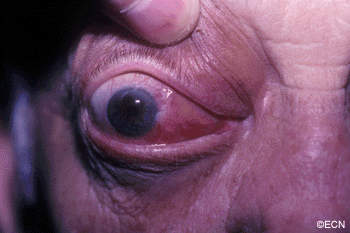
Inflammations can affect the tissues around the eye (orbit and adnexa). Certain orbital inflammations can look like tumors and are therefore called orbital pseudotumor. Orbital pseudotumor can affect one or both eyes of relatively young patients (less than 50 years old). They are not cancer.
Symptoms
Orbital pseudotumor can be quite painful. In fact, pain is one of the most prominent characteristics of this disease. In addition to pain, an inflammatory mass (tumor) can make the patient’s eye protrude (proptosis) and restrict the movement of the eye. A biopsy (called an orbitotomy) is commonly performed to confirm the diagnosis of orbital pseudotumor and to obtain tissue for pathology examination.
Diagnosis
Orbital pseudotumor is typically characterized by the rapid development of pain, proptosis, and swelling around the eye and orbit in adults. Ultrasound and computed tomographic (CT) scanning typically shows a diffuse infiltration of the orbit, an inflammation of the eye wall (sclera), and/or T-sign (with the optic nerve). Orbital pseudotumor related orbital masses typically have poorly defined margins. Systemic testing (blood and spinal fluid) may show signs of inflammation (e.g. increased sedimentation-rate) or atypical cells.
Patients with classic findings of orbital pseudotumor may be treated without a biopsy. A rapid complete response to steroid therapy helps confirm the diagnosis. Atypical cases of orbital pseudotumor usually undergo a diagnostic biopsy.
Specimens can be sent to test for infectious causes of orbital inflammation and certain systemic diseases. Typically eye cancer specialists will obtain blood, skin and radiographic (e.g. x-ray, MRI) tests for a variety of diseases such as sarcoidosis, tuberculosis, and Wegener’s Granulomatosis (see table below). An orbital biopsy can be particularly helpful in diagnosing many of these disorders.
Treatments
Orbital pseudotumor will respond rapidly to high-dose steroid therapy. Unfortunately, when the steroids are stopped, the inflammation often returns. Eye cancer specialists must reduce the steroid medication very slowly in order to prevent recurrence (return) of the disease.
In certain cases, chemotherapy (e.g. methotrexate, cyclosporine) and low-dose radiation (e.g. 1500-2500 cGy EBRT) may be needed to control the inflammation related to orbital pseudotumor. Most patients do well with steroid therapy but they are always at risk for recurrent orbital pseudotumor.
Additional info
Other Common Causes of Orbital Swelling and Inflammation:
"Very well treated by Dr. Finger. He explained everything I needed to know about my issue with detail and attention, putting me at ease and giving me confidence to handle this problem for the rest of my life.”
– N.N.