Paul T Finger, MD (Senior Instructor) Committee Chair, The Ophthalmic Oncology Task Force The American Joint Committee on Cancer- International Union Against Cancer (AJCC-UICC) Instructors: Darryl Ainbinder, MD, Sarah Coupland, MD, James C. Fleming, MD, J. William Harbour, MD, Leonard M. Holbach, MD, Zeynel Karcioglu, MD, Tero Kivelä MD, Hugh McGowan, MD, A. Linn Murphree, MD, Jack Rootman, MD, Stefan Seregard, MD, Valerie White, MD, Christian Wittekind, MD, Goupei Yu, Ph.D.
Language is defined as the human use of spoken or written words as a communication system. Sharing a common language allows us to communicate our ideas and enables progress.
With this in mind, the American Joint Committee on Cancer (AJCC), the International Union Against Cancer (UICC), and the American College of Surgeons (ACS) have come together to sponsor a committee to design a clinically useful TNM (Tumor-Node-Metastasis) based classification for ocular tumors. The AJCC classification system can be used as a universal language for those who diagnose and treat ocular tumors.
Chaired by Paul T. Finger, MD, the AJCC-UICC committee of physicians included specialists in ophthalmic pathology, ocular plastic surgery, intraocular, orbital, and adnexal tumors. Dr. Finger “Internationalized” the committee as to include members from the USA and 10 from other countries.
These included members of the UICC and others with particular expertise based on a review of the published literature. The training of committee members includes: clinical ophthalmic oncology, ophthalmic pathology, molecular biology, genetics, retinal surgery, orbital surgery and biostatistics. In a peer-review process, over 38 reviewers have been involved shaping this work.
All specialists were familiar with current methods of diagnosis and treatment. Thus, every attempt was made to create a clinically useful TNM-based classification system. TNM also means that these classifications conform to what is used throughout medicine, for other tumors and in tumor registries around the world. Clearly, the use of the AJCC-UICC TNM classification would bring ophthalmic oncology into the mainstream of cancer research.
Like Esperanto (created by Dr. L.L. Zamenhof in 1887 to function as an international language), the AJCC-UICC classification is meant to function as a universal eye cancer classification “language” system.1 Esperanto means hopeful. Clearly, we are hopeful that all scientists, clinicians and journals adopt this classification.
Why should we all adopt a single classification system for ocular tumors?
Until we all use the same classifications of ocular tumors, our studies are not directly comparable and the accuracy of meta-analysis is degraded. If we all decide to “speak ocular tumor,” we will embrace an opportunity to allow for accurate comparisons of our investigational studies.
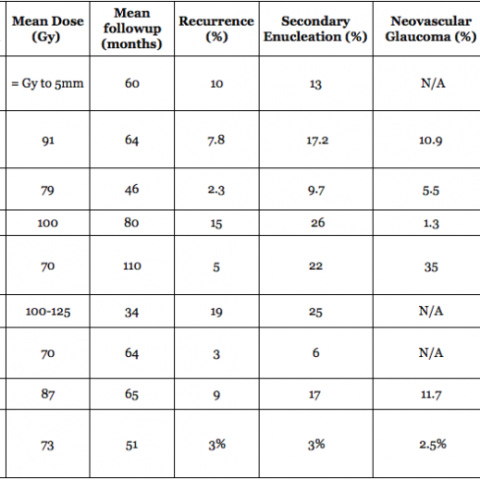
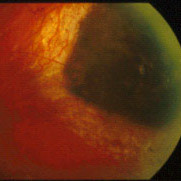
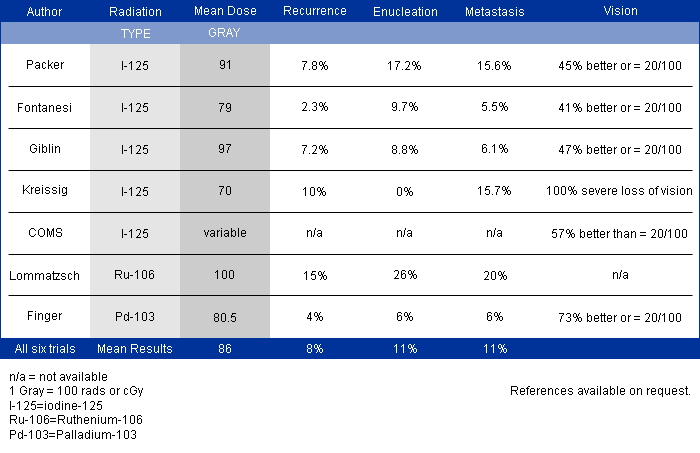
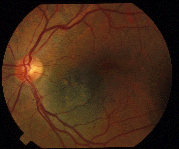
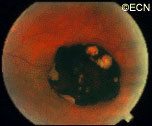
In 1997, Dr. Finger’s review of the literature on “Radiation Therapy for Choroidal Melanoma,” pointed out that no two centers (outside the Collaborative Ocular Melanoma Study [COMS]) published using the same tumor classification.2 It was not possible to state that tumors of equivalent size and location were irradiated at each center.
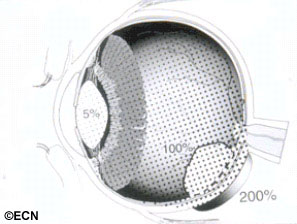
Though important information was obtained by comparing multiple phase-I clinical studies, if all the investigators had used the same tumor-classification-language, it would have improved this comparative analysis. It was not until the COMS that over 40 centers used the same definitions (classifications) of choroidal melanoma size, methods to document location, and radiation dosimetry.3 In fact, the COMS is an excellent example of how a staging system can be used at multiple institutions to allow treatments to be directly comparable and additive. But, the COMS only studied choroidal melanomas. In contrast, the AJCC classification provides clinically useful definitions of tumor size, location and metastatic disease for almost all ocular cancers.4
Why is it important to compare research studies?
We all know that the wheels of progress grind exceedingly slow, but having a common ocular tumor language is an opportunity to grease that wheel. If we all use a common classification for ocular tumors, we will be able to compare treatments on equivalently sized and “staged” tumors. Such information has the potential to improve both our research and clinical decisions.
For example, it would not be reasonable to compare the use of topical mitomycin chemotherapy for multifocal conjunctival and corneal melanoma vs. another study that recruited mostly localized bulbar conjunctival tumors.
Advocates for the AJCC-UICC classification suggest that when a researcher publishes on new treatment for conjunctival melanoma, the reader should be able to discern the relative size, location, and distribution of the tumors treated. Thus, independent clinical studies can be found to be either additive or dissimilar. Our clinical and research decisions are based on ideas and studies and what we have heard in lectures or read in the literature. Clearly, these decisions are only as good as the information we acquire.
Can speaking “Ocular Tumor” affect informed consent?
Absolutely! Much of our time as clinicians is spent explaining the current knowledge and ophthalmic practice to our patients. A typical explanation of the risks and benefits includes what has been proven by statistics-based-research and what is offered as traditional practice. For example, the Reese-Ellsworth classification categorized the size, location and distribution of retinoblastoma in order to prognosticate the effect of external beam radiation therapy.5 Similarly, the AJCC-UICC tumor classifications will allow us to tell our patients that tumors of certain sizes and/or locations are more or less likely to respond to treatments and/or exhibit side effects.
How can we get everyone to employ this common language?
Unlike Esperanto, the AJCC-UICC classification should become mandatory for peer-reviewed publications. This could be done through the instructions for authors (as have other structural elements). We also request that our scientific organizations (e.g. AAO, ARVO, ISER, ASOPRS, ISOO and others) require that scientific data be submitted in the AJCC format. This may sound a bit drastic, but the editors of the major journals and officers of our organizations could make this decision to the benefit of our profession. We also request that our hospitals require that all tumors be AJCC-UICC staged prior to surgery. This would simplify the task of tumor registries all over the world. The data from tumor registries can be used to monitor both the incidence and prevalence of eye cancers.
We hope that peer reviewed journals, web sites, organizations and lay publications support this effort. If the world-wide ophthalmic community learns how to speak and publish in “Ocular Tumor,” we will better understand each other’s work and more effectively help our patients with eye cancer.
Message From the Editor
Reprinted (in part) from Ophthalmology, Vol. 110, Issue 1, 2003, pp 13-14
copyright (2003) with permission from Elsevier Science.
References
- Zamenhof LL. Fundamento de Esperanto, 9th ed. Marmande, France: Esperantaj Francaj Eldonoj, 1963.
- Finger PT. Radiation therapy for choroidal melanoma. Surv Ophthalmol 1997;42:215–32.
- The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, II: characteristics of patients enrolled and not enrolled. COMS Report No. 17. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 2001;119:951–65.
- Haik B, Ainbinder DJ, Finger PT, et al. Part XI: Ophthalmic sites. In: Greene FL, Page DL, Fleming ID, et al., eds. AJCC Cancer Staging Manual, 6th ed. New York: Springer, 2002:347–84.
- Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol Otolaryngol 1963;67:164–72.
Primary Authors:
COL. Darryl Ainbinder, MD (USA)
Sarah E Coupland, MBBS. Ph.D. (UK)
James “Chris” Fleming, MD (USA)
J. William Harbour, MD (USA)
Leonard M Holbach, MD (UICC) (Germany)
Zeynel Karcioglu, MD (USA)
Tero Kivela, MD (Finland)
Hugh McGowan, MD (Canada)
A Linn Murphree, MD (USA)
Jack Rootman, MD (Canada)
Stefan Seregard, MD (Sweden)
Valerie White, B.Sc., MD (Nfld), FRCPC (Canada)
Christian Wittekind, MD (UICC) (Germany)
Guopei Yu, Ph.D. (USA – Statistician)
Reviewers/Participants:
Daniel Albert, MD (USA)
James O. Armitage, MD (USA)
James J. Augsburger, MD (USA)
Nikolas Bechrakis, MD ( Germany)
John H. Boden, MD (USA)
Patricia Chevez-Barrios, MD (USA)
Bertil Damato, MD (UK)
Didi de Wolff-Rouendaal, MD, PhD (The Netherlands)
Laurence Desjardins, MD (France)
Ralph Eagle, MD (USA)
Deepak Edward, MD (USA)
Bita Esmaeli, MD (USA)
Brenda Gallie, MD (Canada)
Dan Gombos, MD (USA)
Jean-Daniel Grange, MD (France)
Hans Grossniklaus, MD (USA)
Barrett Haik, MD (USA)
COL John Halligan, MD (USA)
George Harocopos, MD (USA)
John Hungerford, MD (UK)
Martine Jager, MD (The Netherlands)
Emma Kujala, MD (Finland)
COL Robert A. Mazzoli, MD (USA)
Tatyana Milman, MD (USA)
Tim Murray, MD (USA)
Andrew Schachat, MD (USA)
Arun Singh, MD (USA)
Matthew Wilson, MD (USA)
Section Editor:
Frederick Greene, MD
7th Edition AJCC Cancer Staging Manual Editors
Edge SE, Byrd DR, Carducci MA, Compton CA
AJCC-UICC Administration:
Ms. Karen Pollitt
Ms. Donna Gress
Publisher
Springer, New York, USA
Chair
Paul T. Finger, MD, FACS
The AJCC-UICC Ophthalmic Oncology Task Force
Tel: 1212-832-8170
Related Links
Search Google for Do You Speak Ocular Tumor? The 11/10/2008 AAO Course in Atlanta