“Think of sunglasses as sunblock for your eyes,” is an oft-repeated slogan around the New York Eye Cancer Center. While most people tend to think of sunglasses as just a fashion statement, they may actually be a key defense against eye cancer. Sun causes damage to eye tissues just like it does to skin – but since you can’t put sunblock on your eyeballs, you should make sure to wear UV-cancelling sunglasses as well.
Fortunately, other groups are joining in this educational effort.
Ocular melanoma is the most common primary eye tumor in adults. There are around 2,000 new cases every year. These tumors are closely related to skin cancer. Since exposure to the sun is a known risk factor for skin cancer, it follows that UV rays from the sun may also increase the risk of developing this form of eye cancer. There is circumstantial evidence linking exposure to sunlight and ocular melanoma. This form of eye cancer is more common in patients with blue eyes, and who work in outdoor occupations.
When any form of light is absorbed by the body, a reaction occurs resulting in heat and chemical changes. Ultraviolet light rays are particularly energetic and cause more chemical reactions in ocular tissues than visible light. This means more potential damage to the eye.
While the link between sunlight and eye melanomas is not conclusively proven, it is worth a small precaution that will make you look like James Dean anyway!
A good pair of sunglasses will block all UVA, UVB, and UVC rays. You want to make sure the glasses provide 100% UV protection. Many optical shop have a machine called a photometer that measures UV transmission through glasses. You want to wear sunglasses that block all UV radiation or light under 400 nm in wavelength.
Buyer beware: Shades that don’t offer this protection – or cheap pairs that fraudulently claim to – may actually increase the risk of eye cancer by allowing more UV light into the eye.
So choose wisely, make that fashion statement, and protect your eyes at the same time.
The following is a statement from The Eye Cancer Foundation, reprinted here with their permission:
The Eye Cancer Foundation (ECF) today announces a bold new strategic direction with a focus on bringing patients in every country access to an #EyeCancerCure. This will be accomplished through the Eye Cancer Working Day events, sponsored fellowships to train specialists in underserved countries, and the Bioinformatics Grid (BIG) project. The ECF seeks to broaden participation in its efforts, facilitate information sharing, and promote more rigorous research practices.
To this end, the Foundation has revamped its website at the new domain of eyecancercure.com, launching a truly international effort to achieve an #EyeCancerCure for as many patients as possible. Typing “eyecancercure” will now take you to the Foundation’s presence on Facebook, Twitter, and the web.
This new strategic vision reflects both a focusing of scope and a widening of participation. The Eye Cancer Foundation got its start some two decades ago through the efforts of our Chairman Dr. Paul T. Finger to fill the need for an online resource with essential information for eye cancer patients and physicians. The result was The Eye Cancer Network, which has grown and remained essential reading for anyone affected by or treating eye cancer. In the meantime, the ECF built a Board of committed benefactors, a truly global network of eye cancer specialists, and a brigade of passionate volunteers. Together, we have accomplished many important projects, especially developing an international, scientific, AJCC-UICC language used to describe most types of eye cancers.
Today, the ECF has reached a point that we are ready to diversify participation and focus our mission. Dr. Finger generously furnished the vast majority of content on The Eye Cancer Network. He will henceforth be personally developing this trove of information into an “online textbook” for continued use by patients and practitioners for years to come. Meanwhile, the new Foundation website highlights our key projects, inspiration and support for patients, and the world’s largest eye cancer specialist directory. In the coming years, we will be reaching out to eye cancer specialists around the world to provide new community-based content contributed by our many affiliated physicians and patient members. Dr. Finger remains dedicated to his role as Chairman of The Eye Cancer Foundation Board as we seek more contributors to accomplish the ambitious goals we have set for the next 5 years.
Are you ready to achieve an #EyeCancerCure? Now is the time to get involved. Contact us for more information.
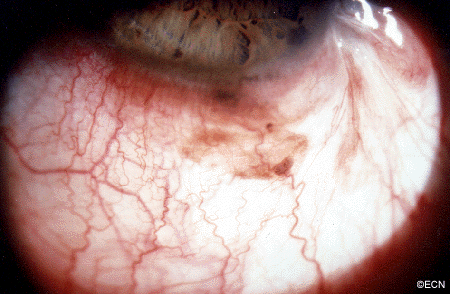
External photograph shows the pigmented tumor “melanoma” in the conjunctiva.
By Paul T. Finger, MD
Current treatments for conjunctival cancer has included surgical removal, removal with subsequent cryo-(freezing)-therapy, radiation therapy, and chemotherapy eye-drops. While most treatments have focused on avoiding large surgeries which may (in very severe cases) be associated with vision loss or loss of the eye, these decisions have been made with the knowledge that conjunctival cancers are serious. They can invade into the orbit (tissues around the eye), the sinuses, and the brain. Conjunctival melanomas and squamous carcinomas can also spread (metastasize) to other parts of the body. Because of high recurrence rates after standard treatments and the desire to avoid surgery, recent investigations have focused on topical chemotherapy “eye-drops.”
For a more detailed explanation of the risks and potential benefits of topical chemotherapy for conjunctival cancers, the editor suggests you obtain and read the referenced manuscripts.
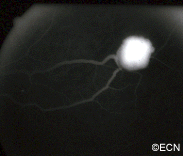
After 28 days of topical MMC chemotherapy the tumor regressed.
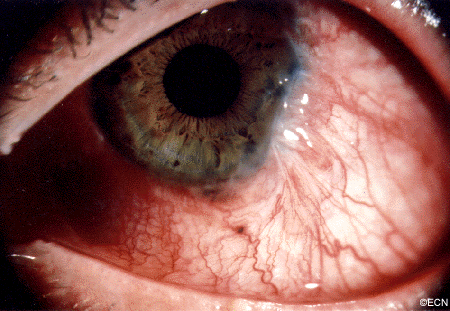
Conjunctival Melanoma and PAM with Atypia
In 1993, Dr. Finger was the first to use mitomycin chemotherapy eye-drops to treat conjunctival melanoma and primary acquired melanosis. Since that time, it has become commonly and widely used throughout the world. It is currently used to reduce the tumor prior to surgical removal and with surgery for cases where there is too much normal tissue involved (too much for surgery and/or freezing (cryotherapy).
In addition, Dr. Finger discovered that topical interferon chemotherapy (Intron A) eye drops can be used to treat superficial conjunctival melanoma.
Topical chemotherapy eye drops can be used as primary treatment, as well as an addition to surgery and cryotherapy. Since almost all conjunctival tumors have different sizes, shapes and locations, your eye cancer specialist should know when and if you are a candidate for topical chemotherapy.
Squamous Conjunctival Neoplasia
Chemotherapy eye-drops have also been investigated as an alternative treatment of squamous conjunctival – corneal neoplasia. For more information consider reading the following manuscripts. Squamous Conjunctival Neoplasia
References
Finger PT, Milner MS, McCormick SA. Topical chemotherapy for conjunctival melanoma. British Journal of Ophthalmology 77:751-3, 1993
Frucht-Pery J, Pe’er J. Use of mitomycin C in the treatment of conjunctival primary acquired melanosis with atypia. The Archives of Ophthalmology 1996;114:1261-1264.
Finger PT, Czechonska G, Liarikos S. Topical mitomycin C chemotherapy for conjunctival melanoma and PAM with atypia British Journal of Ophthalmology 82:476-9, 1998
Finger PT, Sedeek RW, Chin KJ. Topical interferon alfa in the treatment of conjunctival melanoma and primary acquired melanosis complex. Am J Ophthalmol. 2008 Jan;145(1):124-129.
Danapoulos ED, Danpoulous IE, Liarikos SB et al. Effects of urea treatment in malignancies of the conjunctiva and cornea. Ophthalmologica 178:198-203, 1979
De Keizer RJW, de Wolff-Rouendaal, van Delft JL, et al. Topical application of 5-FU in premalignant lesions of the cornea, conjunctiva, and eye lid. Doc Ophthalmol 64:31-42, 1986.
Frucht-Pery J, Rozenman Y. Mitomycin C treatment for conjunctival -corneal intraepithelial neoplasia: A multicenter experience. Ophthalmology 104:2085-2093, 1997
Wilson MW, Hungerford JL, George SM, Madreperla SA. Topical mitomycin C for the treatment of conjunctival and corneal epithelial neoplasia. American Journal of Ophthalmology 124:303-311, 1997
The authors examined if the American Joint Committee on Cancer (AJCC) staging system for ocular adnexal lymphoma (OAL) could be used to predict local control and systemic disease. A multicenter, consecutive case series of patients with biopsy-proven conjunctival, orbit, eyelid, or lacrimal gland/sac lymphoma was performed.
Results
They found that extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue was the most common and AJCC clinical stages were cT1NOMO (21.7%), cT2NOMO (44.6%), cT3N0M0 (5%), and cT4NOMO (2.4%).
Treatment was able to control the local tumor in 75% of patients.
Of the 19 local recurrences, 74% (most) were found after non-radiation based treatments.
The lower-risk “smaller tumor” AJCC T1 and T2 groups without lymph node involvement or metastatic disease had longer disease-free survival than the higher-risk AJCC T1, T2 with nodal involvement or metastatic disease, AJCC T3, and T4 as well as Ann Arbor II, III, and IV.
Conclusions
Regardless of stage, recurrence and disease-free survival were more closely related to having had radiation treatment and histopathology rather than site specific factors, such as tumor size or location around or behind the eye.
Q: Can the laser be used to treat an eye melanoma?
A: Most studies have shown that the laser treatment just burns the surface of the melanoma. This can leave live cancer cells under the tumor’s laser scarred surface and in the wall of the eye. It may be just a matter of time before the cancer will grow again. If the cancer is not killed or removed, there is a chance that it might spread to the rest of your body.
Due to the high incidence of failure using Transpupillary Thermotherapy (TTT) laser alone, it is rarely used without additional plaque radiation for choroidal melanoma. However, intraocular hemorrhages are common, and in rare cases the wall of eye (sclera) can be weakened allowing tumor growth into the tissues behind the eye.
In contrast, TTT laser is commonly successful in treatment of small retinoblastomas.
Q: Can you surgically remove the tumor and leave my eye?
A: During this type of surgery the eye must be opened. Though not a proven risk for metastasis, eye wall resection allows tumor cells to float out of the eye and into the orbit (spaces around the eye).
With lamellar (split thickness) eye wall resection, the wall of the eye (sclera) is sewn back in place. The COMS study showed that up to 50% of choroidal melanomas had invaded the wall of the eye. Therefore, with lamellar eye wall resection, the entire tumor is not always removed or treated. In order to prevent recurrence, eye cancer specialists are currently adding plaque therapy to treat the eye wall. Lastly, depending on the skill of the operating surgeon, many eyes do not tolerate eye wall resection. These eyes suffer sight limiting retinal detachment, cataract, and intraocular hemorrhage.
Q: How long will I have to be a patient?
A: For the rest of your life. You must be followed by an eye-care specialist and may need a medical oncologist. You may want to consider this when choosing your doctors and their location.
Patients who receive plaque radiotherapy are typically seen by their eye care specialist every 3 to 4 months. Patients may undergo twice-yearly blood tests, radiographic imaging studies and physical examinations. Patients being treated for radiation retinopathy and/or optic neuropathy may have to be seen every 4-6 weeks.
Copies of your laboratory evaluations should be forwarded to your eye cancer specialist’s office, so they can be double-checked for metastatic disease. In the United States, the law requires that you must request this in writing from your family doctor, internist, or oncologist.
Q: How will having a choroidal melanoma affect my life?
A: Almost all patients who have been determined to have an ocular tumor (e.g. choroidal melanoma) are treated and then return to their normal activities.
The time between the diagnosis and the completion of treatment can be very stressful. Rely on your doctors and family for help. After you are treated they should encourage you to get back to your normal activities. Just remember to be periodically monitored by your eye care specialists and general medical doctors.
Q: What are the standard treatment options?
A: Standard treatments for choroidal melanoma are enucleation and radiation.
Enucleation means removal of the eye. In most cases, it is the only form of treatment that allows your doctor to completely remove the tumor from your body. Unfortunately, patients also lose their vision and the cosmetic use of the eye. With time, almost all patients are able to do all the things they used to before losing their eye. Since the COMS study found no difference in survival between enucleation and plaque radiation therapy (for medium-sized melanomas), few doctors will recommend enucleation if “eye-sparing” radiation therapy is possible.
The most common alternative to removal of the eye is radiation therapy, and the most commonly used radiation technique for intraocular melanoma is eye-plaque radiation therapy.
Plaque technique involves placing a dish-shaped plaque-device (about 1 to 2 centimeters in diameter) on the outer surface of the eye, under the tumor. The plaque is implanted behind the eye, left in place for up to 5-7 days, and then removed. During treatment patients are likely to notice no more than occasional discomfort.
The effects of radiation on the tumor are typically measured 3 to 4 months after treatment. Eventually, eye melanomas shrink to about 40% of their pretreatment size. Though they rarely disappear, these tumors are considered to be dead. In most studies, irradiated tumors will regrow (within the eye) in less than 8% of cases. Local control means the tumor was killed. Ask your doctor about their rates of local control after treatment and for how long those patients were followed (typically reported at 5 and 10 years after treatment). You can see The New York Eye Cancer Center’s local control rates on our outcomes page.
Note about radiation: More than 95% of patients have no problems associated with plaque surgery. As with any ocular surgery, there can be secondary retinal detachments, hemorrhages, or infections. There are also the regular risks related to hemorrhage, infection or reaction to anesthesia. Most plaque patients do not develop these problems.
Q: What research is being done for ocular melanoma?
A: Around the world, researchers are working on new methods of diagnosis and treatment for choroidal melanoma, retinoblastoma, and other ocular tumors. One only has to do a PubMed search to find thousands of published articles.
It is important to point out two major research initiatives:
The first multi-center, multinational prospective randomized clinical study carried out for an eye cancer was called The Collaborative Ocular Melanoma Study (COMS). These types of studies offer the best evidence-based statistically significant information currently available. The National Eye Institute (NEI) and the National Institute of Health (NIH) supported the COMS.
The COMS had three studies:
The Small Choroidal Melanoma Study confirmed that orange pigmentation, leakage of fluid, and thickness are all correlated to tumor growth and metastatic potential. This study also found that there exists a risk of metastatic melanoma from small choroidal melanomas.
The Medium-Sized Choroidal Melanoma Study determined that plaque radiation was equal to removal of the eye for the prevention of metastatic choroidal melanoma.
The Large-Sized Choroidal Melanoma Study determined that 2000 cGy of external radiation prior to enucleation does not prevent the spread of choroidal melanoma.
The second major collaborative step forward was when The American Joint Committee on Cancer joined with the International Union Against Cancer to develop a Tumor-Node-Metastasis (TNM) grading system for most eye cancers. It is our hope that this common language will enable centers (around the world) to compare their results.
Q: What usually happens to a patient’s vision after treatment?
A: Enucleation
After enucleation (removal of the eye) there is no vision from that eye. The patient is considered monocular and sees from the remaining eye. Most patients see well from their remaining eye and live normal lives. Loss of one eye does affect depth perception, but with time, most patients are able to adjust. Consider that part of depth perception comes from the relative size of objects (distant objects appear smaller).
A: Plaque Radiation Therapy
Prior to treatment for radiation-related damage to the retina and optic nerve, post-plaque radiation therapy for choroidal melanoma caused vision loss in half of patients within 5 years. However, with the use of laser treatment and/or intraocular anti-VEGF drug therapy, the vision destroying radiation retinopathy can be suppressed. Anti-VEGF suppression of radiation retinopathy and optic neuropathy has preserved patient vision for years. Further, the natural eye looks and moves better than an ocular prosthesis.
Disadvantages of plaque versus enucleation include that you must see an ophthalmologist every 3 to 4 months after treatment for dilated ophthalmoscopy and ultrasound measurements of your tumor. This is because patients who have their eye irradiated (plaque or proton) develop radiation complications within the eye, and there is a small chance that the tumor might regrow.
The patient’s chance for functional vision after radiation is related to how close the tumor is to the functional center of the retina (macula), the type of radiation, and the dose delivered to normal ocular structures. The American Brachytherapy Society Consensus Guidelines suggest that all centers perform pre-operative comparisons of plaque types for dose to normal eye structures.
Q: Will the tumor spread to other parts of my body?
A: Less than 4% of patients are found to have metastatic melanoma at the time of diagnosis. But with time after treatment, a much larger percentage are found to develop metastasis. This difference is thought to be due to undetectable microscopic melanoma cells present at the time of treatment. It is not helpful to dwell on percentages, but your doctor should be able to give your approximate chance of developing metastasis based on your tumor’s size and location.
About Enucleation Surgery
Q: If I must lose my eye, will it hurt?
A: The eye is surrounded by bones; therefore, it is much easier to tolerate removal of an eye as compared to loss of a lung or kidney.
Almost all enucleation surgery can be performed as an outpatient. Older patients and those with significant systemic medical problems may have to stay in the hospital after surgery.
Since this surgery is usually performed under general anesthesia, the patient does not feel or see anything during surgery. Long-acting local anesthesia (6 hours) can be given during surgery, allowing for the least amount of pain possible (when you wake up in the recovery room). Most patients who have their eye removed have a mild headache for 24-36 hours after surgery.
Q: What is enucleation?
A: Enucleation is removal of the eye. It is the form of treatment that allows your eye cancer specialist to remove the tumor from your body. Unfortunately, patients also lose all the vision and the cosmetic use of the eye. With time, almost all patients are able to do most all the things they used to do before their eye was removed.
Q: What might I look like after enucleation?
A: This is a patient who has completed cosmetic rehabilitation after enucleation surgery. Notice that he looks normal, but the prosthetic eye does not move as well as a normal eye.
Q: When will I get a prosthetic eye?
A: Patients can usually have a temporary prosthesis (that looks like an eye) within 10 days of enucleation surgery. Besides the swelling and the “black-eye,” you will look fairly normal. After a final prosthetic fitting, 90% of patients are happy with the way they look, and 80% say others can’t even tell they are monocular.
Read the book A Singular View, by Frank Brady. It will help in your transition. This book was written by an airplane pilot who lost one eye. We suggest that you wear unbreakable polycarbonate glasses to protect your good eye.
Q: Why are very large-sized tumors treated by removal of the eye?
A: This is because the amount of radiation required to kill a large tumor that fills most of the eye is just too much for the eye to stand. Within months to years, many patients who are treated with radiation for very large ocular melanomas lose vision, develop glaucoma, or have their eye removed anyway. It is important that the eye cancer specialist inform each patient regarding their approximate chance of developing radiation associated complications. Despite these risks, more and more patients with large intraocular tumors are treated with eye and vision-sparing radiation therapy.
Frequently Asked Questions After Enucleation Surgery
Q: How long will I have to be a patient?
A: For the rest of your life. You must be followed by an eye-care specialist and may need a medical oncologist. You may want to take this into account in choosing your doctors and their location.
An eye care specialist examines patients who are enucleated twice a year. In addition, patients should have blood tests, radiographic imaging studies, and physical examinations. Copies of your laboratory evaluations should be forwarded to your eye-cancer specialist’s office so they can be double-checked for metastatic disease. The law requires that you must request this in writing from your family doctor, internist, or oncologist.
Q: How often will I need to be checked after enucleation?
A: Dr. Finger recommends that you have an eye examination within 1 week, 1 month, and at least every 6 months after surgery. This is to monitor for inflammation or infection, and because there is an extremely small chance the tumor will regrow behind your prosthesis.
Dr. Finger recommends that you return for a complete ophthalmic oncology examination at least on a yearly basis. You should also have twice-yearly medical check-ups by your family doctor, internist, or medical oncologist.
Q: What sort of care will my eye need after I go home?
A: You will be advised to take topical antibiotic and steroid medications for one month. These medicines will help you heal more safely and quickly.
For the first week after surgery, you will tear a lot. These tears may contain a little blood. This is normal. Once a day, you should gently wash the outside of your eyelid with a warm, clean, soapy washcloth. Don’t let matter accumulate to form a crust on your eyelids.
Q: What will happen to my vision?
A: Enucleation means removal of the eye. It is a form of treatment that completely removes the primary tumor from the body, but the patient loses all vision and the cosmetic use of the eye. It takes some time to adjust to using one eye, but almost all patients learn to compensate within the first year after surgery.
Q: When can I get a prosthesis?
A: Dr. Finger can usually place a temporary prosthesis (that looks like an eye) within 10 days of enucleation surgery. Most patients are fitted with a permanent prosthesis 4 to 6 weeks later.
Q: When can I go back to normal activity?
A: Do not lift, strain, or rub your eye for at least 14 days after surgery.
Do not take aspirin or other blood thinners unless your internist says it is required. The orbit should heal quickly, and you should be able to return to school or work within 2 to 6 weeks after surgery.
Q: When can I wash my hair and face?
A: You should gently clean your lids each day. You may use a clean wash cloth and baby shampoo. You should not rub your eyelids or run the shower at your operated eye for at least 10 days after surgery.
How do I choose an Eye Cancer Specialist?
Q: How do I pick a doctor?
A: You should pick a doctor who has experience caring for eye cancer patients and with whom you are comfortable. You should be able to ask questions about your case and feel comfortable that your treatment decisions are not only based on available science, but also take into account your personal situation and medical problems. Since you may need to see your doctors for many years, consider that it is easier to be treated and followed with a doctor in your area.
Consider the following:
You should be guaranteed to see the doctor with whom you are making the appointment (not a substitute doctor). Ask when making your appointment.
Make sure that your surgeries will be performed by the doctor you want. Ask the doctor who will be performing your surgery when signing your consent form.
You should be able to ask questions about your case and treatment decisions.
Ask how easy it is to speak with your doctor before and after surgery.
Ask what happens when there is an emergency during nights and weekends.
Make sure your doctor has the expertise to manage any eye and/or systemic problems you may have in the future.
Q: How long will I have to be a patient?
A: For the rest of your life. You must be followed by an eye cancer specialist and may need a medical oncologist. You may want to take this into account when choosing your doctors and their location. Patients who receive plaque radiotherapy are typically seen every 3-4 months. Patients who are enucleated are usually seen twice a year. Many eyelid and orbital tumor patients are advised to follow up with their eye cancer specialist, at least, on a yearly basis.
In any case, your eye care specialist should check to make sure that you have regularly scheduled medical workups, including an evaluation for metastatic disease (as necessary). Copies of your laboratory evaluations should be forwarded to your eye-cancer specialist’s office so that they can be double-checked. The law requires that you must request this in writing from your family doctor, internist, or oncologist.
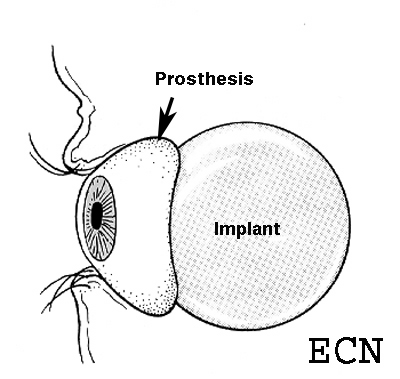
Enucleation: About Ocular Prosthesis Care
Ocular Prosthesis
Q: How do I remove my prosthesis?
1) First, wash your hands.
2) Then you should place a towel over your lap or sink to act as a net for the prosthesis if it slips out of your hand. Should it fall it could scratch, break, or get lost.
Manual Technique
3) Place one finger on the temporal (towards the ear) aspect of the lower lid on top of the cheek bone.
4) Look up.
5) Cup your other hand under your eye (to catch the prosthesis).
6) Gently press your finger in and pull the eyelid skin towards your ear (on that side).
7) The edge of your prosthesis will likely be emerging at the edge of the lower eyelid, or (less likely) it has fallen into your cupped hand.
8) If the prosthesis is just barely out, you can use a finger on your other hand to rotate it out of the socket.
9) Don’t be surprised if some discharge comes along with the prosthesis.
Suction Technique
1) Hard contact lens suction devices are commercially available in drug stores and vision centers.
2) These devices can be squeezed to create a vacuum that attaches the device to the front of the prosthesis.
3) Once attached, the patient can lift the bottom portion of the prosthesis out from beneath the lower lid, then slide the superior portion down towards the cheek.
Once the Prosthesis Is Out
1) Commercially available sterile saline solution should be used to clean your eye socket.
2) Now you can consider cleaning the prosthesis.
Q: How do I clean my prosthesis?
1) Place the prosthetic eye into a container that can be filled with liquid as to cover the prosthesis.
2) Full or half strength hydrogen peroxide solution can be used to soak the prosthesis for 10 to 15 minutes. After soaking, remove the prosthesis from the container and rinse it with sterile saline solution.
3) Prosthesis cleaning is typically performed once or twice a week (as instructed by your eye care professionals).
4) Continuous and consistent periodic cleaning of the prosthesis will increase your comfort, decrease secretions, prevent secondary conjunctivitis and extend the life of your ocular prosthesis.
Q: How often do I need to have my prosthesis professionally cleaned or replaced?
1) You should return to your ocularist for professional cleaning and polishing 2 times each year.
2) Most patients get a new prosthesis every 3 to 5 years because even with excellent maintenance, the tissues around the prosthesis can change and the artificial eye can become scratched.
Warning!!!
If you notice excessive discharge, swelling or irritation, you should call your eye care professional immediately.
Related Links
For the more medically minded, you can go to a medical library, or click and order a copy of our comprehensive review of:
A: No! Once the radiation plaque is removed, all the radioactivity is gone. There will be no radiation left in your body, your clothing, your glasses, or any of your personal belongings. However, the effects of the radiation delivered to your tumor and eye will continue to be observed for months and years after treatment.
Q: How often will I need to be checked after plaque treatment?
A: We recommend that you be examined every 3-4 months after plaque radiation. This is because there is a small chance the tumor will regrow or that your eye may develop radiation-related problems (that may require treatment).
In addition to seeing your local eye care specialist, we recommend that you return for a complete ophthalmic oncology examination (at least) on a yearly basis.
You should also have twice-yearly medical check-ups by your family doctor, internist, or medical oncologist. Dr. Finger recommends that you have a medical checkup at least two times a year. This should include a physical examination and radiographic imaging of the abdomen.
Q: How quickly will my tumor go away after plaque radiation?
A: Tumors are usually measured to shrink after 3 months. We don’t measure a tumor before three months, because they can swell and become temporarily larger after radiation, unnecessarily frightening patients. Since the goal of radiation therapy is to prevent the tumor from growing, don’t be concerned if your tumor shrinks slowly. Most tumors eventually shrink to about 40% of their original size and rarely disappear. A residual lump of dark, shrunken tumor typically persists for years after treatment.
Q: How soon can I return to normal activities?
A: The eye should heal quickly. You should not lift any objects greater than 10 pounds or rub your eye for at least two weeks after surgery. Then you can return to normal activity. Patients usually return to school or work within 2-4 weeks after completion of treatment. If the tumor has caused a retinal detachment, it may take longer for you to return to normal activities.
Q: What after-care will my eye need after I go home?
A: After ophthalmic plaque radiation therapy, the patient is usually requested to take eye-drops daily for about a month. These drops contain antibiotics, steroids, and medicines to relax your eye. These drops help your eye heal more safely and quickly. For the first week after surgery, you will tear and the tears may contain a little blood. This is normal. You should gently wash the outside of your eyelid with a warm, clean, soapy wash cloth. Don’t let matter accumulate to form crust on your eyelids.
Q: What are the differences between Protons versus Plaques?
A: Proton Beam Radiotherapy is a form of external beam irradiation. It involves directing radiation through the front of the eye, lids, and orbit in order to reach the intraocular tumor. Eyelash loss, eyelid excoriation, corneal neovascularization and ulceration, dry eye, neovascular glaucoma, and cataract have been reported to be more common after proton beam radiation therapy.
Proton beam radiation therapy typically requires surgical clips to be sewn onto the eye (around the tumor) this helps the radiation therapist to direct the beam into the eye. Eye movements are monitored on a video screen because if the eye moves, the beam moves away from the tumor. If the eye is detected to move, the radiation technologist will temporarily turn off the beam until the eye is repositioned. In contrast, radiation eye plaques are sewn onto the eye as to cover the base of the intraocular tumor. So when the eye moves, so does the plaque. With eye plaque, eye movement is not thought to appreciably affect the distribution of radiation within the eye.
Both proton beam and Fingers’ slotted plaques can be used to treat certain tumors that are near, touch or even surround the optic nerve.
Eye-Plaque Radiotherapy typically involves attaching a dish-shaped radiation source beneath the tumor and leaving it there for 5-7 days. With proton beam, once the marking clips are surgically placed, the patient visits the radiation center for 3-5 treatments. The marker-clips are not typically removed.
Compared with Proton-Beam, the front of the eye usually receives much less radiation with plaque radiation therapy, but parts of the retina may receive more. This is why anterior “front of the eye” complications are much less commonly reported after low-energy ophthalmic plaque radiation therapy.
Q: What is a radioactive eye-plaque?
A: A radioactive eye-plaque is a device that can be used to deliver a high-dose of radiation to an intraocular tumor.
This is because when an eye-plaque contains radioactive iodine-125 or palladium-103 seeds, the gold of the plaque blocks more than 99% of radiation. Therefore, when the plaque is sewn to the outside of your eye (underneath the tumor), the radiation is directed into the eye. For an average-sized tumor, less than 10% of the radiation makes it out of the other side of the eye.
Plaques come in various sizes (diameters between 10 to 22 mm). A plaque will be chosen to cover your entire tumor plus at least a 2-millimeter “safety margin”. The extra area is included to make sure the entire tumor is within the targeted area. The plaque is implanted utilizing several standard techniques (transillumination, ophthalmoscopy and ultrasound). Intraoperative ultrasound imaging and light-assisted plaque localization are techniques developed to make plaque-placement more accurate (see innovations).
Radiation treatment is continuous and will typically take up to 7 days. At the end of treatment, your tumor will have been given all the necessary radiation. Once your plaque is removed, there will be no radioactivity left in your body.
Radioactive plaques come in various sizes with suture eyelets so that it can be temporarily attached to the wall of the eye for treatment.
Q: What is plaque radiation therapy?
A: Radioactive plaque therapy is a form of treatment, which allows the eye-cancer specialist to destroy your tumor without removing the eye. Unfortunately, the radiation can also affect the normal parts of your eye and harm your vision. Your ophthalmic and radiation oncologists will work together to try to increase the effectiveness of radiation (to kill your cancer), while decreasing the chance that radiation will harm the normal parts of your eye.
Q: What is proton beam radiation therapy?
A: Protons are charged nuclear particles that can be sent into the eye. These particles travel through and are somewhat absorbed by tissues on the way to your intraocular tumor. The targeted zone (the tumor and surrounding 2-3 mm) gets most of the radiation. The proton radiation field is in the shape of a tube, with little side-scatter or dose posterior to the eye. Like plaque radiotherapy, the amount of radiation to the normal parts of the eye depends of the size and location of the tumor.
Q: What radiation precautions must be taken?
A: While the radiation-plaque is sewn onto your eye, certain precautions must be observed. These rules will ensure that visitors and patients in surrounding areas do not receive radiation exposure in excess of the Nuclear Regulatory Commission (NRC) regulations. A list of rules are given to you before you go home with the plaque in place and can be disregarded after you have the radiation-plaque removed.
Q: What will happen to my vision?
A: Radiation may cause eventual blurring, dimming, or rarely a total loss of vision (in the eye with the tumor). The amount of vision loss depends on what your vision was before treatment, how close the tumor is to the center of your retina (the fovea), and how sensitive your tissues are to radiation. Most plaque-irradiated eyes maintain some central vision, and almost all retain peripheral vision. Plaque radiation should not affect the vision in the other eye.
Q: Why does the plaque stay in place for up to 7 days?
A: Plaque radiation therapy is delivered over to a dose known to destroy the cancer, and at a dose rate that will be tolerated by the eye. The rate at which the radiation is delivered is affected by both the radiation source and the size of the tumor.
In consultation with the radiation oncologists, eye plaques are individually designed and constructed for each patient. Specialized computer programs are used to calculate the total dosage and speed of radiation. The precise distribution of radiation throughout the eye is calculated and used to determine the risks of secondary radiation complications.
Q: Will my hair fall out from radiation?
A: It is normal for patients to have fears about radiation.
The type of radiation used in most eye plaque therapy should not cause hair loss, nausea, brain damage, or affect your other eye.
Other forms of radiation therapy can cause hair loss within the field of irradiation. For example, proton beam radiation therapy is associated with eyelash loss. External beam therapy can also cause hair loss where it enters and exits the head.
Q: Should I Get a Second Opinion?
A: Second opinions are great when all your doctors say the same thing. Unfortunately, differing opinions can make things even more confusing. Remember to ask the doctor:
What are the standard treatment options for my tumor?
If the proposed treatment is “new” or investigational? If so, how many patients have been treated? How long have they been followed for recurrent disease?
What have your doctor’s results been for tumor control and vision retention?
Is the proposed treatment FDA or CE approved?
Who will be the operating surgeon? In the case of plaque therapy; who will put it in and who will take it out?
Does your center compare radiation plaque types (before surgery) to determine which one would be best for my tumor?
Does your center keep plaque patients in the hospital or send them home during treatment and why?
Does your eye cancer specialist and center personally follow treated patients for recurrence and metastatic disease?
How soon will you ask me to return after surgery?
What tests do you perform (over time) to monitor my body for metastatic disease?
What does your eye cancer specialist do if I am found to have metastatic disease?
What About Retinoblastoma?
Q: Until what age can a child develop another retinoblastoma?
A: Most retinoblastomas are diagnosed before the age of 3. Most eye cancer specialists agree that a child with retinoblastoma is not likely to develop a new tumor after 7 years of age.
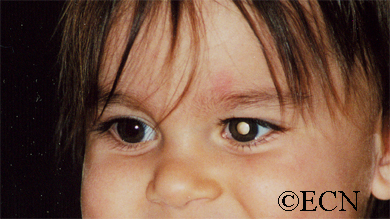
Q: What are the most common symptoms of retinoblastoma?
A: Most children are found to have a white pupil “leukocoria,” or a deviated eye “strabismus,” or secondary painful glaucoma. In less developed countries patient more commonly present with orbital or optic nerve extension or metastasis.
Q: What other eye diseases can cause leukocoria “white pupil?”
A: Other than retinoblastoma, cataract, Coat’s disease, persistent hyperplastic primary vitreous, and retinopathy of prematurity can cause the appearance of a white pupil.
Q: Who gets retinoblastoma?
A: There are typically 325 of new cases of retinoblastoma diagnosed each year in North America. Ten thousand cases are estimated to occur throughout the world. Retinoblastoma occurs equally in boys and girls, different ethnic groups, and in either eye.
Retinoblastoma is a genetic cancer. Therefore, 10% of patients will have a family history of retinoblastoma, and 25% will affect both eyes. Forty percent of patients have the retinoblastoma gene throughout their body. The abnormal gene is 13q14.2, that is chromosome 13, long arm (q), band 14.2. Though it is the most common primary intraocular cancer in children, intraocular leukemia is more common.
Von Hippel-Lindau (VHL) disease is an autosomal dominantly inherited multisystem cancer syndrome with a predilection for the central nervous system (CNS) and the retina. Retinal capillary “Vvon Hippel” hemangioma is one of the most common and often the earliest manifestations of VHL disease and, therefore, ophthalmologists are frequently involved in the care of patients with this disease. The incidence of VHL disease is approximately one in 40,000 live births and it is estimated that there are approximately 7,000 patients with VHL disease in the United States.
Symptoms
Ophthalmic Findings:
The main ophthalmic finding in VHL is retinal capillary hemangioma, which is a benign hamartoma. The anterior segment can be secondarily involved due to complications such as neovascular glaucoma and cataract formation. A large cohort study found only 2% of eyes had neovascularization of the iris. If a patient has a solitary retinal capillary hemangioma, they do not necessarily have VHL disease. However, they should undergo genetic testing.
Diagnosis
Retinal capillary hemangiomas are usually orange red, circumscribed, round, vascular tumors supplied by a pair of dilated and tortuous feeder vessels. They are most commonly located in the temporal peripheral retina. Juxtapapillary retinal capillary hemangiomas are less common, representing about 11% to 15% of cases, and their appearance can vary depending on whether the lesion is endophytic (grow from the retina into the vitreous gel of the eye), exophytic (grow beneath the retina), or sessile (flat).
Retinal capillary hemangioma usually manifests as a solitary tumor, but approximately one-third of patients have multiple retinal hemangiomas, and up to half of patients have bilateral involvement.
Secondary effects (from the retinal angioma) leading to visual loss, such as intraretinal and subretinal exudation, are often limited to the vicinity of the hemangioma but can be remote, often producing a macular star of exudates. Glial proliferation can lead to tractional retinal detachment and macular pucker. Retinal or vitreal hemorrhages are rarely observed, occurring in fewer than 3% of cases.
Frequency and Patient Related Findings:
The frequency of occurrence of retinal capillary hemangiomas in VHL disease has been
reported to vary from 49% to 85%. The mean age at diagnosis of retinal capillary hemangioma in VHL disease is approximately 25 years, and most patients present between the ages of 10 and 40 years. The probability of developing a retinal capillary hemangiomas increases progressively with age. Recent publications indicate that the hemangioma is usually manifested by age 30, and the prevalence rate is stable thereafter. Therefore adults with a normal retina at age 30 years may have a low risk of developing a retinal capillary hemangioma during the reminder of their lives. The natural course of retinal capillary hemangiomas is variable (progression, stability or spontaneous regression). Small lesions may remain stable for years or may show evidence of gliosis without leakage, but some have been documented to enlarge. Most hemangiomas, however, tend to enlarge progressively and lead to retinal changes. In late stages they may cause massive exudation and retinal detachment, uveitis (inflammation), glaucoma and phthisis (shrinking of the eye). Classification systems to aid in staging the clinical progression have been developed.
Other Tumors That May Look Like Von Hippel Angiomas:
The fundus findings of retinal capillary hemangioma are usually typical, and diagnosis can be made based on ophthalmoscopic examination. The diagnosis might be confused with retinal macroaneurysm or adult Coats disease when severe exudation exists.
Treatments
For treatment of Von Hippel retinal angiomas, see Conditions (Von Hippel Angioma) However, in ths section we will discuss the diagnosis of systemic disease and genetics:
The systemic manifestations of VHL are multiple and include CNS hemangiomas of the brain and spinal cord, renal cell carcinomas, renal cysts, pheochromocytomas, pancreatic cysts, islet cell tumors, epididymal cystadenomas, endolymphatic sac tumors of the inner ear, and adnexal papillary cystadenomas of the broad ligament. After retinal capillary hemangioma, the most frequently affected organ systems are the CNS, kidneys and adrenal glands, many of them occurring years after the initial presentation with retinal capillary hemangiomas.
The diagnosis of VHL disease is based on three elements which include:
retinal capillary hemangioma or CNS hemangioma
visceral lesions
family history of similar lesions
Surveillance:
After diagnosis is made, screening protocols should be followed, including urinary catecholamines and ophthalmoscopy on an annual basis with MRI of the brain and spinal cord every 2 to 3 years, and yearly abdominal US with an additional abdominal CT scan every 2 to 3 years.
Genetic Testing:
VHL disease is an autosomal dominant disease whose gene is located on chromosome 3p 25-26. The gene functions as a tumor suppressor gene that promotes tumor formation when its function is lost. The normal protein product of the VHL gene forms a complex with other proteins that targets hypoxia inducible factors (HIFs) for degradation. Mutations in the VHL gene result in stabilization of the HIFs, which bind to specific enhancer elements in the VEGF gene and stimulate angiogenesis. With a near-complete penetrance of the disease and only rare instances of mosaicism, genetic testing has been proved helpful in early diagnosis and clinical screening for disease manifestations.
SUMMARY
VHL disease is an autosomal dominantly inherited multisystem cancer syndrome with a predilection for the central nervous system and the retina. Retinal capillary hemangioma is one of the most common and earliest manifestations of VHL disease. Fundus findings are usually typical, and diagnosis can be made based on ophthalmoscopic examination, but fluorescein angiography is an additional informative diagnostic tool. Various treatment modalities exist, although the mainstays of therapy are laser photocoagulation and cryotherapy. VHL disease, however, is associated with significant mortality secondary to either CNS hemangioma or renal cell carcinoma. Life expectancy of affected individuals can be improved by early detection, genetic testing and systemic treatment. Treatment of this syndrome requires cooperation of physicians from multiple specialists, including those to treat the central nervous system and kidney tumors.
References
Annesly WJ, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:446-453.
Choyke PL, Glenn GM, Walther et al. The natural history of renal lesions in von Hippel- Lindau disease: a serial CT study in 28 patients. Am J Roentgenol. 1992;159:1229-1234.
Dahr SS, Cusick M, Roudriguez-Coleman H, et al. Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina. 2007;27:150-158.
Harris AL. von Hippel-Lindau syndrome. Target for anti-vascular endothelial growth factor (VEGF) receptor therapy. The Oncologist. 2000;5(suppl):32-36.
Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943-944.
Magee MA, Kroll AJ, Lou PL, Ryan EA. Retinal capillary hemangiomas and von Hippel-Lindau disease. Semin Opthalmol. 2006;21:143-150.
Maher ER, Yates JR, Harries, et al. Clinical features and natural history of von Hippel-Lindau disease. QJM. 1990;77:1151-1163.
Maher ER, Lselius L, Yates JR, Et al. von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443-447.
Shields CL, Shields JA, Barret J, et al. Vasoproliferative tumors of the ocular fundus. Classification and clinical manifestations in 103 patients. Arch Ophthalmol. 1995;113:615-623.
Sigelman J. Retinal diseases. Pathogenesis, laser therapy and surgery. Boston:Little Brown and Company. 1984:316.
Vail D. Angiomatosis retinae, eleven years after diathermy coagulation. Am J Ophthalmol. 1958;46:525-534.
Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371-378.
Wong WT, Chew EY. Ocular von Hippel-Lindau disease: clinical update and emerging treatments. Curr Opin Ophthalmol. 2008;19:213-217.
Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology. 2008;115:1957-1964.
Dr. Finger is a Principal Investigator in the Collaborative Ocular Melanoma Study Group
Collaborative Ocular Melanoma Study Publications: Peer-review papers only.
1. Collaborative Ocular Melanoma Study Group: Accuracy of diagnosis of choroidal melanomas in the Collaborative Ocular Melanoma Study. COMS Report No. 1. Arch Ophthalmol 108:1268-1273, 1990.
2. Collaborative Ocular Melanoma Study Group: Complications of enucleation surgery. COMS Report No. 2. In: Proceedings of the Symposium on Retina and Vitreous (Rudolph M. Franklin, ed.). New Orleans Academy of Ophthalmology. Kugler Publications, New York, 1993; pp. 181-190.
3. Collaborative Ocular Melanoma Study Group: Design and methods of a clinical trial for a rare condition: The Collaborative Ocular Melanoma Study. COMS Report No. 3. Controlled Clin Trials 14:362-391, 1993.
4. Collaborative Ocular Melanoma Study Group: Mortality in patients with small choroidal melanoma. COMS Report No. 4. Arch Ophthalmol 115:886-893, 1997.
5. Collaborative Ocular Melanoma Study Group: Factors predictive of growth and treatment of small choroidal melanoma. COMS Report No. 5. Arch Ophthalmol 115:1537-1544, 1997.
6. Collaborative Ocular Melanoma Study Group: Histopathologic characteristics of uveal melanomas in eyes enucleated from the Collaborative Ocular Melanoma Study. COMS Report No. 6. Am J Ophthalmol 125:745-766, 1998.
7. Collaborative Ocular Melanoma Study Group: Sociodemographic and clinical predictors of participation in two randomized trials: Findings from the Collaborative Ocular Melanoma Study. COMS Report No. 7. Controlled Clin Trials 22:526-537, 2001.
8. Grossniklaus HE, Albert DM, Green WR, Conway BP, Hovland KR for the Collaborative Ocular Melanoma Study Group: Clear cell differentiation in choroidal melanoma. COMS Report No. 8. Arch Ophthalmol 115:894-898, 1997.
9. Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma. I: Characteristics of patients enrolled and not enrolled. COMS Report No. 9. Am J Ophthalmol 125:767-778,1998.
10. Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma. II. Initial mortality findings. COMS Report No. 10. Am J Ophthalmol 125:779-796,1998.
11. Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma. III. Local complications and observations following enucleation. COMS Report No. 11. Am J Ophthalmol 126:362-372, 1998.
12. Collaborative Ocular Melanoma Study Group: Echography (ultrasound) procedures for the Collaborative Ocular Melanoma Study. COMS Report No. 12. J Ophth Nurs Technol Part I, 18(4):143-149, Part II, 18(5):219-232, 1999.
13. Collaborative Ocular Melanoma Study Group: Consistency of observations from echograms made centrally in the Collaborative Ocular Melanoma Study. COMS Report No. 13. Ophthalmic Epidemiol 9:11-27, 2002.
14. Collaborative Ocular Melanoma Study Group: Cause-specific mortality coding: Methods in the Collaborative Ocular Melanoma Study. COMS Report No. 14. Control Clin Trials 22: 248-262, 2001.
15. Collaborative Ocular Melanoma Study Group: Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study. COMS Report No. 15. Arch Ophthalmol 119:670-676, 2001.
16. Collaborative Ocular Melanoma Study Group: Collaborative Ocular Melanoma Study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years. COMS Report No. 16. Ophthalmology 108(2):348-366, 2001.
17. Collaborative Ocular Melanoma Study Group: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma. II. Characteristics of patients enrolled and not enrolled. COMS Report No. 17. Arch Ophthalmol 119: 951-965, 2001.
18. Collaborative Ocular Melanoma Study Group: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma. III. Initial mortality findings. COMS Report No. 18. Arch Ophthalmol 119: 969-982, 2001.
19. Collaborative Ocular Melanoma Study Group: The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS Report No. 19. Ophthalmology 198:2197-2206, 2002.
20. Collaborative Ocular Melanoma Study Group: Trends in size and treatment of recently diagnosed choroidal melanoma, 1987-1997. Findings from patients evaluated at Collaborative Ocular Melanoma Study centers. COMS Report No. 20. Arch Ophthalmol 121:1156-1162, 2003.
21. Collaborative Ocular Melanoma Study Group: Comparison of clinical, echographic, and histologic measurements from eyes with medium-sized choroidal melanoma in the Collaborative Ocular Melanoma Study. COMS Report No. 21. Arch Opthalmol 121:1163-1171, 2003.
22. Collaborative Ocular Melanoma Study Group: Ten-year follow-up of fellow eyes of patients enrolled in Collaborative Ocular Melanoma Study (COMS) randomized trials. COMS Report No. 22. Ophthalmology 111:996-976, 2004.
23. Diener-West M, Reynolds SM, Agugliaro DJ, Caldwell R, Cumming K, Earle JD, Green DL, Hawkins BS, Hayman I, Jaiyesimi I, Kirkwood JM, Koh W-J, Robertson DM, Shaw JM, Thoma J. Screening for metastasis from choroidal melanoma: Experience of the Collaborative Ocular Melanoma Study. Collaborative Ocular Melanoma Study Report No. 23. Am J Clin Oncol 22:2438-2444, 2004.
24. Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma. IV. Ten-year mortality findings and prognostic factors. COMS Report No. 24. Am J Ophthalmol 138:936-951, 2004.
25. Melia BM, Moy CS, McCaffrey L: Quality of life in patients with choroidal melanoma: A pilot study. Ophthalmic Epidemiol 6:19-28, 1999.
26. COMS Quality of Life Study Group: Quality of life assessment in the Collaborative Ocular Melanoma Study: Design and methods. COMS-QOLS Report No. 1. Ophthalmic Epidemiol 6:5-17, 1999.
27. COMS Quality of Life Study Group: Development and validation of disease-specific measures for choroidal melanoma. COMS-QOLS Report No. 2. Arch Ophthalmol 121:1010-1020, 2003.
28. Collaborative Ocular Melanoma Study Quality of Life Study Group. Quality of life after iodine 125 brachytherapy versus enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study prospective study. COMS-QOLS Report No. 3. Arch Ophthalmol (under revision for resubmission, July 2004).
29. Collaborative Ocular Melanoma Study Group: The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma. IV. Ten-year Mortality findings and prognostic factors. COMS Report No. 24. Am J Ophthalmol 138:936-951, 2004.
30. Collaborative Ocular Melanoma Study Group: Second Primary Cancers after Enrollment in the COMS Trials for Treatment of Choroidal Melanoma. COMS Report No. 25. Archives of Ophthalmology 2005:123:601-4.
Von Hippel angioma can grow within the retina or optic nerve. They characteristically have a “feeding” retinal arteriole and a “draining” retinal vein. Bilateral involvement can be seen in up to 50% of individuals.
Von Hippel angioma are vascular tumors, not cancers and do not metastasize. Twenty percent of patients will be found to have the von Hippel Lindau Syndrome–associated with cerebellar hemangioma, pheochromocytomas, visceral cysts and renal cell carcinomas.
Symptoms
Von Hippel angioma patients either have no symptoms, or become symptomatic due to secondary retinal detachment or rarely neovascular glaucoma. The symptoms of retinal detachment are flashes of light, spots in the vision (floaters), and loss of vision. The symptoms of neovascular glaucoma are eye pain, light sensitivity, vision loss, and headache.
Diagnosis
Some patients with von Hippel angioma will have a family history of this disease. Von Hippel angioma are usually visible by dilated eye examination (ophthalmoscopy). Ultrasound can be used to measure the tumor’s size, and to evaluate for high internal reflectivity. Ophthalmoscopy typically reveals a dilated feeder artery and draining vein. An associated retinal detachment may be seen around the tumor or may be so large as to cover (obscure) an underlying von Hippel angioma.
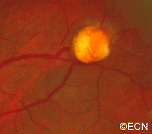
Fluorescein Angiography – Note the feeder and draining vessels, as well as the bright “light-bulb” appearance.
Fluorescein Angiography: Eye-care specialists perform studies of the blood vessels in the eye with a synthetic organic dye called fluorescein. The dye is injected into the arm and travels to the blood vessels inside the eye. If a tumor is in the eye, we can see specific characteristics of its circulation which can help us differentiate between it and other types of tumors. Von Hippel angiomas have a unique pattern of circulation with a feeder arteriole and a draining vein. Since the tumor extends from the retina into the eye (vitreous humor), von Hippel angiomas exhibit intense hyperfluorescence, often compared to a “light-bulb.”
Treatments
Von Hippel angiomas can appear in both an autosomal dominant hereditary or sporadic forms. All patients should be given periodic systemic examinations including imaging studies for cerebellar hemangiomas and renal cell carcinoma. Family members should be examined with indirect ophthalmoscopy. Genetic testing is available (see related links below).
The treatment of retinal capillary hemangiomas can be a challenge to the ophthalmologist due to the presence of bilateral multiple tumors and the likelihood of new tumor formation. Despite treatment, up to 25% of cases can have permanent visual loss of acuity less to than 20/40 in one or both eyes. Various treatment modalities including observation, cryotherapy, plaque radiotherapy, and vitreoretinal surgery have been utilized.
Recent advances in the understanding of VHL protein function and tumorigenesis have led to new treatments targeting the biology of the disease, as opposed to ablative or surgical approaches. Molecules upregulated or increased in the context of a VHL mutation, such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), have been targeted in investigational anti-angiogenic therapies, both in systemic manifestations of the disease and in ocular disease.
OBSERVATION AS TREATMENT
Observation is rarely employed due to the tendency of retinal capillary hemangiomas to progress. However, observation only might be chosen in small (Juxtapapillary hemangiomas (those next to the optic nerve disc) are particularly difficult to treat and are initially managed with observation because they can remain stable for years. As a general rule; since these tumors are not cancer and cannot metastasize; treatment should only be undertaken in case of tumor progression or a threat to visual acuity due to the adverse effect of treatment on the optic nerve and major blood vessels.
LASER PHOTOCOAGULATION
Laser photocoagulation is currently used to treat small retinal capillary hemangiomas located in the retina in eyes with clear media. When possible, we first occlude the feeder artery, then (if necessary) to surround/demarcate the posterior 180 degrees of the tumor, lastly and again if needed directly treat the tumor’s surface. Patients should be informed that multiple laser treatment sessions are typically required. Potential complications include retinal detachment, retinal and vitreous hemorrhages.
CRYOTHERAPY
Typical indications for cryotherapy are anterior retinal location of the hemangioma and massive subretinal fluid, which can reduce the laser energy uptake. Double freeze-thaw technique is employed under indirect ophthalmoscopic observation. A 15-year review found that most all hemangiomas under 3.75 mm in diameter successfully responded to cryotherapy.
ANTI-VEGF STRATEGIES
Recent studies have indicated that anti-VEGF strategies may be effective. However, no large clinical trials have been reported.
Additional Info
Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology. 2008;115:1957-1964.
Annesly WJ, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:446-453.
Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943-944.
Small melanomas can be watched for growth prior to treatment. Should growth occur, then the patient knows the melanoma will eventually destroy the vision and increase the chance that cancer cells will spread to other parts of the body. The Collaborative Ocular Melanoma Study (COMS) was interested in how many small melanomas would grow and over what period of time. The COMS found that more than 25% of small melanomas were found to grow (within 2 years of follow-up). Since choroidal melanoma growth is the best predictor for vision loss and increased risk of metastasis, this COMS finding underscores the need to follow patients with small melanomas closely after diagnosis.
Medium-sized Choroidal Melanoma Study
The medium-sized tumor study was designed to determine if iodine-125 plaque-irradiation is better, equal, or worse than enucleation (removal of the eye) for the prevention of metastasis. In this study, half of enrolled patients were treated by enucleation and the other half underwent plaque radiation therapy. Patients were followed for evidence of recurrence and metastatic melanoma.
Half of enrolled patients were treated by enucleation and the other half underwent plaque radiation therapy. Patients were followed for evidence of recurrence and metastatic melanoma.
The COMS medium-tumor trial concluded that there is no significant difference between these two treatment options with respect to survival. COMS centers had followed 80% of patients for at least 5 years at the time they issued their report. Therefore, COMS found no evidence that removing the eye is a better treatment than iodine-125 plaque radiation therapy for preventing spread of choroidal melanomas.
Large-sized Choroidal Melanoma Study
Large-melanoma trial was designed to see if radiation before enucleation (removal of the eye) would prevent metastasis. The idea was to see if pre-operative irradiation would sterilize any cells that might break free during surgery. The other half of the patients did not receive radiation before their surgery.
The Large-sized Choroidal Melanoma Study concluded that patients who received 2000 rads (cGy) of external irradiation to their eye before it was removed, had an equal chance of developing metastatic disease as compared to those who were treated by enucleation (removal of the eye) alone.
Retinoblastoma is the most common intraocular cancer of childhood and affects approximately 300 children in the United States each year. More than 96% of children in North America and Europe are cured of retinoblastoma due to early detection and treatment of the affected eye. This is not true for children in countries that do not have eye cancer specialists.
Unfortunately, some children can have both eyes affected. Whenever possible, eye-cancer specialists try to save a child’s eye and preserve their vision.
Symptoms
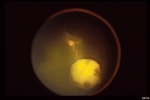
An intraocular photograph of an isolated “endophytic” retinoblastoma.
Leukocoria (white pupil) and misaligned eyes (strabismus) are the most common signs of retinoblastoma. In other cases, the child may have developed neovascular glaucoma and may be in pain. Longstanding glaucoma can cause enlargement of the eye (buphthalmos). Children with neovascular glaucoma and enlargement of the eye are at greater risk for extraocular spread of their retinoblastoma.
A family history of retinoblastoma can be very important. Retinoblastoma was the first cancer to be directly associated with a genetic abnormality (Deletions or mutation of the q14 band of chromosome 13). Retinoblastoma can occur sporadically (without a family history) or it can be inherited (with a family history).
If a genetic mutation is found there is a 45-50% chance that the parents will have another child with retinoblastoma. If there is no family history and no mutation is found, the risk of having a second child with retinoblastoma is 2-5%. The average age of children first diagnosed with retinoblastoma is 18 months (typical range 0 to 36 months).
Diagnosis
More than 75% of children with retinoblastoma are first noted to have a “white-pupil” (which the doctors call leukocoria), or poorly aligned eyes (which the doctors call strabismus), or a red and painful eye (usually due to glaucoma). Other eye diseases which can cause these symptoms include congenital cataract, Toxocara canis, Coat’s disease, and persistent hypertrophic primary vitreous (PHPV). These diseases may look like retinoblastoma, but by performing an eye examination under anesthesia (EUA), specialized blood tests, digital photography, radiographic scans, and ultrasound evaluations ophthalmic oncologists can diagnose intraocular retinoblastoma in over 95% of cases. In order to be 100% correct all the time, eye-cancer specialists would have to perform a biopsy. Biopsies of intraocular retinoblastoma are avoided in order to prevent cancer cells from spreading outside of the eye.
The presence of orbital extension, uveal involvement, and optic nerve invasion are known risk factors for the development of metastatic retinoblastoma.
Treatments
Retinoblastoma treatment typically requires the cooperation of an ophthalmic oncologist, pediatric oncologist, and radiation therapist. Over the last 30 years, treatment has evolved from simple enucleation (removal of the eye), to eye-sparing radiotherapy, and more recently to chemotherapy-based multi-modality therapy (for selected cases). Intra-arterial chemotherapy (IAC) has recently been investigated to save eyes, vision and spare the child from systemic chemotherapy.
Though retinoblastoma has been cured by external beam irradiation, investigators have found that radiation may cause an increase in the risk of developing second cancers later in life.
Protocols are currently being evaluated to use chemotherapy to shrink the retinoblastoma in order to treat them with laser therapy, freezing therapy (cryotherapy), and local “plaque” radiation. Where applicable, these techniques are thought to be safer than external beam irradiation for retinoblastoma. Intra-arterial chemotherapy is a newer method of perfusing the eye with chemotherapy, used for selected cases.
Treatment of retinoblastoma often requires a team of doctors made up of ophthalmic, radiation and pediatric oncologists. These doctors should evaluate your child, discuss all the different forms of treatment, and make them available.
"Very well treated by Dr. Finger. He explained everything I needed to know about my issue with detail and attention, putting me at ease and giving me confidence to handle this problem for the rest of my life.”
– N.N.