Von Hippel Angioma
Description
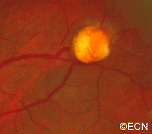
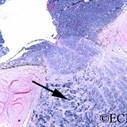
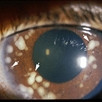
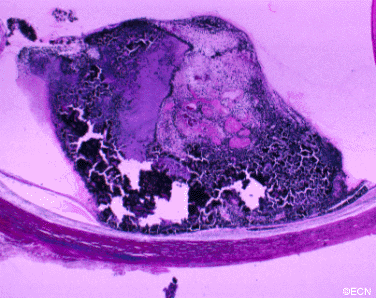
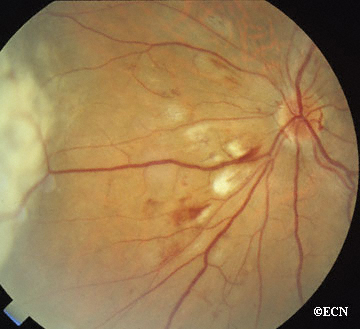
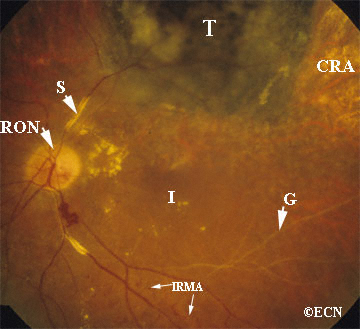
Von Hippel angioma can grow within the retina or optic nerve. They characteristically have a “feeding” retinal arteriole and a “draining” retinal vein. Bilateral involvement can be seen in up to 50% of individuals.
Von Hippel angioma are vascular tumors, not cancers and do not metastasize. Twenty percent of patients will be found to have the von Hippel Lindau Syndrome–associated with cerebellar hemangioma, pheochromocytomas, visceral cysts and renal cell carcinomas.
Symptoms
Von Hippel angioma patients either have no symptoms, or become symptomatic due to secondary retinal detachment or rarely neovascular glaucoma. The symptoms of retinal detachment are flashes of light, spots in the vision (floaters), and loss of vision. The symptoms of neovascular glaucoma are eye pain, light sensitivity, vision loss, and headache.
Diagnosis
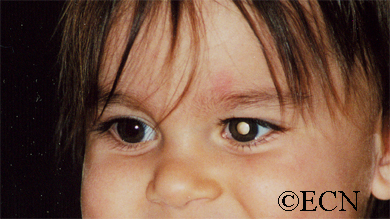
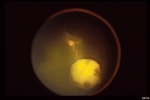
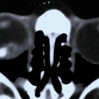
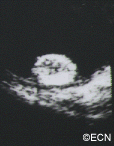
Some patients with von Hippel angioma will have a family history of this disease. Von Hippel angioma are usually visible by dilated eye examination (ophthalmoscopy). Ultrasound can be used to measure the tumor’s size, and to evaluate for high internal reflectivity. Ophthalmoscopy typically reveals a dilated feeder artery and draining vein. An associated retinal detachment may be seen around the tumor or may be so large as to cover (obscure) an underlying von Hippel angioma.
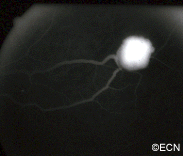
Fluorescein Angiography: Eye-care specialists perform studies of the blood vessels in the eye with a synthetic organic dye called fluorescein. The dye is injected into the arm and travels to the blood vessels inside the eye. If a tumor is in the eye, we can see specific characteristics of its circulation which can help us differentiate between it and other types of tumors. Von Hippel angiomas have a unique pattern of circulation with a feeder arteriole and a draining vein. Since the tumor extends from the retina into the eye (vitreous humor), von Hippel angiomas exhibit intense hyperfluorescence, often compared to a “light-bulb.”
Treatments
Von Hippel angiomas can appear in both an autosomal dominant hereditary or sporadic forms. All patients should be given periodic systemic examinations including imaging studies for cerebellar hemangiomas and renal cell carcinoma. Family members should be examined with indirect ophthalmoscopy. Genetic testing is available (see related links below).
The treatment of retinal capillary hemangiomas can be a challenge to the ophthalmologist due to the presence of bilateral multiple tumors and the likelihood of new tumor formation. Despite treatment, up to 25% of cases can have permanent visual loss of acuity less to than 20/40 in one or both eyes. Various treatment modalities including observation, cryotherapy, plaque radiotherapy, and vitreoretinal surgery have been utilized.
Recent advances in the understanding of VHL protein function and tumorigenesis have led to new treatments targeting the biology of the disease, as opposed to ablative or surgical approaches. Molecules upregulated or increased in the context of a VHL mutation, such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), have been targeted in investigational anti-angiogenic therapies, both in systemic manifestations of the disease and in ocular disease.
OBSERVATION AS TREATMENT
Observation is rarely employed due to the tendency of retinal capillary hemangiomas to progress. However, observation only might be chosen in small (Juxtapapillary hemangiomas (those next to the optic nerve disc) are particularly difficult to treat and are initially managed with observation because they can remain stable for years. As a general rule; since these tumors are not cancer and cannot metastasize; treatment should only be undertaken in case of tumor progression or a threat to visual acuity due to the adverse effect of treatment on the optic nerve and major blood vessels.
LASER PHOTOCOAGULATION
Laser photocoagulation is currently used to treat small retinal capillary hemangiomas located in the retina in eyes with clear media. When possible, we first occlude the feeder artery, then (if necessary) to surround/demarcate the posterior 180 degrees of the tumor, lastly and again if needed directly treat the tumor’s surface. Patients should be informed that multiple laser treatment sessions are typically required. Potential complications include retinal detachment, retinal and vitreous hemorrhages.
CRYOTHERAPY
Typical indications for cryotherapy are anterior retinal location of the hemangioma and massive subretinal fluid, which can reduce the laser energy uptake. Double freeze-thaw technique is employed under indirect ophthalmoscopic observation. A 15-year review found that most all hemangiomas under 3.75 mm in diameter successfully responded to cryotherapy.
ANTI-VEGF STRATEGIES
Recent studies have indicated that anti-VEGF strategies may be effective. However, no large clinical trials have been reported.
Additional Info
- Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology. 2008;115:1957-1964.
- Annesly WJ, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:446-453.
- Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943-944.